高顺式HTPB 对不饱和树脂的改性
2021-09-01赵亚静李伯耿介素云
赵亚静, 李伯耿, 介素云, 徐 力
(化学工程联合国家重点实验室(浙江大学), 浙江大学 化学工程与生物工程学院, 浙江 杭州 310027)
1 前 言
不饱和聚酯树脂(unsaturated polyester resin,UPR)通常由不饱和聚酯预聚物与活性稀释剂(如苯乙烯)组合而成,其中不饱和聚酯预聚物为不饱和二元酸、饱和二元酸和多元醇的缩聚产物。由UPR 固化形成的复合材料耐化学腐蚀,具有优异的刚性和电性能,加工简便、性价比高,被广泛地用于化工、机械、电器、交通运输和建筑等行业[1],是热固性树脂中用量最大的品种之一。然而,UPR 固化后硬而脆,低温韧性差,限制了在海洋和航空航天等领域中的应用。提高UPR 的抗冲击性具有重要意义。
UPR 的增韧主要有3 条途径:1)共混引入第二相材料弹性体(包括以共固化形式引入的端基含官能团的液体橡胶)[2-7]和刚性颗粒[8-14],弹性体的引入需解决弹性体相尺寸的控制与机械强度的损失问题,刚性颗粒的引入需解决分散与相容性问题;2)采用互穿网络技术[15-24],通常构建互穿网络的工艺复杂、流程长、成本高;3)改进不饱和聚酯预聚物的分子结构,接枝或嵌段引入柔性链如己二酸、聚乙二醇、桐油等[25-29],或选择恰当的交联剂增加交联网链的活动能力[30-33]。该方法未考察低温下不饱和聚酯的柔性改善效果。
近来,本课题组开发了一种顺丁橡胶环氧化—裂解—加氢还原制液态高顺式端羟基聚丁二烯(hydroxyl-terminated polybutadiene,HTPB)的新技术[34-37]。该技术可精确调控HTPB 的相对分子质量,并可确保端羟基的官能度非常接近于2。因此,前期用HTPB 部分替代二元醇,将这种具有超低玻璃转变化温度( ≤ -100 ℃)的遥爪型液体橡胶引入聚氨酯链中,很好地改善了它们低温下的韧性[38-39],拉伸强度也得到提高[39]。本文则研究这种HTPB 对UPR 的改性作用,即利用HTPB 两端的羟基通过共缩聚反应将顺式聚丁二烯低聚物引入UPR 的预聚物链中,进而考察它们的固化物在常温和低温下的力学性能及热性能。
2 实验部分
2.1 主要原料
顺丁烯二酸酐(MA),分析纯;邻苯二甲酸酐(PA),分析纯;1,2-丙二醇(1,2-PG),分析纯;以上均来自阿拉丁生化科技股份有限公司。苯乙烯,分析纯;二甲苯,分析纯;以上均来自国药集团化学试剂有限公司。过氧化环己酮,50% 酞酸二丁酯溶液;环烷酸钴,8% 钴;以上均来自百灵威科技有限公司。各原料使用前均不作任何处理。
高顺式端羟基聚丁二烯,由本课题组自创的顺丁橡胶的环氧化、裂解和还原法制得。由核磁共振氢谱测得摩尔质量为2 065 g·mol-1,邻苯二甲酸酐酯化法测算得羟基官能度为2.02,差示扫描量热仪(DSC)测得玻璃化转变温度为-104.6 ℃。所用的顺丁橡胶聚合原液来自浙江传化合成材料有限公司,制备及分析测试方法见文献[36]。
2.2 样品制备
不饱和聚酯预聚物的制备:采用先酯化后缩聚的两步法,将1,2-丙二醇、顺丁烯二酸酐与邻苯二甲酸酐按物质的量比为1.1:0.5:0.5 加入配有机械搅拌、通气和分水装置的三口烧瓶中,加入二甲苯通过共沸蒸馏除水,通入N2防止聚酯颜色加深及过早凝胶。酯化反应在160~190 ℃进行,每隔0.5 h测定酸值,直至酸值低于80 mg·g-1;拆除冷凝装置,换上带有冷阱、真空表、安全瓶以及真空泵的抽真空装置,保持温度不变减压反应1~2 h,当体系酸值降至50 mg·g-1时停止反应,迅速降温至100 ℃,趁热加入一定量的苯乙烯(质量分数w=40%),搅拌均匀,得到不饱和聚酯预聚物的黏稠液体。
HTPB 改性的不饱和聚酯预聚物(UPR-HTPB)的制备:将本实验室自制的高顺式HTPB 替代部分原料1,2-丙二醇,其余操作不变。制得的改性样品中HTPB 的质量分数为预聚物(不包括苯乙烯)中的1%~5%。合成过程如图1 所示。

图1 UPR-HTPB 预聚物的合成路线Fig.1 Synthetic scheme for UPR-HTPB prepolymer
固化:向不饱和聚酯预聚物中,加入引发剂过氧化环己酮(w=0.5%)和促进剂环烷酸钴(w=1%),搅拌均匀,倒入模具中,室温下固化24 h,再在80 ℃下固化3 h。
2.3 结构表征与性能测试
分别对固化前的不饱和聚酯预聚物和固化后的不饱和树脂样品进行结构表征和性能测试。
不饱和聚酯预聚物样品结构用德国Bruker 公司的Bruker DMX-400 共振仪氢谱(1H-NMR)表征,以氘代氯仿为溶剂,四甲基硅烷为内标。表征前用索氏抽提法,以环己烷为溶剂回流48 h 除去其中微量未反应的HTPB。
不饱和树脂固化样品的性能测试包括室温与低温(-20 ℃)下的拉伸试验、无缺口冲击试验,以及动态力学性能(DMA)分析、热重(TG)分析、冲击试验断裂面形貌观察。拉伸试验按ASTM D 638-91 标准,采用德国Zwick 公司的带有控温箱的Zwick/Roell 万能材料测试机。无缺口冲击试验按ASTM D 256 标准,采用环球(香港)科技有限公司的CEΛST 低温摆锤冲击测试仪。冲击断裂面形貌的观察采用日本日立公司的SU-3500台式扫描电镜(SEM);因不饱和聚酯导电性差,对冲击断裂面喷金,放大1 000 倍。DMA 分析采用美国TA公司的TA-Q800 动态热机械分析仪;测试时,将样品快速降温至-100 ℃并平衡5 min,以5 ℃·min-1的速率升温至160 ℃,固定频率1 Hz,进行三点弯曲试验。TG 分析采用美国TA 公司的TA-Q500 热重分析仪;取3~5 mg 固化后样品置于样品盘,氮气氛下以10 ℃·min-1的速率由室温升至600 ℃进行测试。
3 结果与讨论
3.1 U PR-HTPB 的结构表征
预聚物的1H-NMR 分析结果如图2 所示。谱图中特征峰a~f 可分别归属为图1 中马来酸1,2-丙二醇酯结构单元和邻苯二甲酸1,2-丙二醇酯结构单元中的各─CxHy─基团;特征峰h~m 可分别归属为活性稀释剂苯乙烯中的各─CxHy─基团[40]。化学位移δ= 1.29~1.38 处的特征峰归属为─CH3基团,因HTPB 中不含─CH3基团,故它们的峰强和峰的位置均未发生明显变化。与纯UPR 的图谱相比,可以发现所有的UPR-HTPB 谱图中δ=2.08 附近均出现了新的峰,且随着HTPB 的质量分数增加,峰强增大。这归因于HTPB 中的─CH2基团[36],证明合成了UPR-HTPB。

图2 不同HTPB 质量分数的UPR 的核磁图谱Fig.2 1H-NMR spectra of UPR with different HTPB contents
3.2 拉伸性能
不同HTPB 质量分数的固化UPR 的室温与低温拉伸曲线如图3 所示。从图中可见,无论室温或低温下,HTPB 的引入均可显著提高固化UPR 的断裂伸长率,且随HTPB 质量分数的增大而增大; HTPB的引入还使固化UPR 的拉伸强度显著提升。表明HTPB 在不饱和聚酯预聚物链中的引入既可改善UPR的柔性,又可改善其强度;且低温下这种改性效果尤为明显。

图3 不同HTPB 质量分数的UPR 的应力-应变曲线Fig.3 Stress-strain curves of UPR with different HTPB contents
进一步对拉伸曲线进行分析,由图3(a)可见,室温下纯UPR 的断裂伸长率为(1.7±0.4)%,随着HTPB质量分数的增加,UPR 的断裂伸长率呈上升趋势,当HTPB 的质量分数w(HTPB)<3% 时增速最快,之后逐渐平稳。当w(HTPB)=5% 时,断裂伸长率达最大值(3.8±0.3)%,比纯UPR增大了124%。当低温(-20 ℃)时,由图3(b)可见,纯UPR 的断裂伸长率约为0.5%,明显低于室温下的断裂伸长率。随HTPB 的引入,UPR 的断裂伸长率几乎随HTPB 质量分数的增加呈线性地增大;当w(HTPB)=5% 时,断裂伸长率为(1.6±0.1)%,接近于室温下纯UPR 的断裂伸长率,比低温下纯UPR 的断裂伸长率增大了220%。
由图3(a)可见,室温下纯UPR 的拉伸强度为(36.3±6.7) MPa;当w(HTPB)<4%时,UPR 的拉伸强度随着HTPB 质量分数的增加而增大;当HTPB 质量分数继续增加,拉伸强度有所降低,但仍明显高于纯UPR 的拉伸强度。当w(HTPB)=3%时,室温下的拉伸强度可达(76.8±2.8) MPa,比纯UPR 拉伸强度增大了112%。HTPB 的引入同样可增大低温下UPR 的拉伸强度。由图3(b)可见,低温下纯UPR 的拉伸强度为(15.2±0.8) MPa,不到室温下拉伸强度的一半;引入HTPB 可使UPR 的拉伸强度显著增大,且在w(HTPB)=1%~5% 的实验范围内随HTPB 质量分数的增加而增大;当w(HTPB)=5% 时,低温下的拉伸强度达(43.4±0.9) MPa,比纯UPR 的拉伸强度增大了186%,甚至高于室温下纯UPR 的拉伸强度。弹性体共混改性塑料,在增韧的同时往往使其拉伸强度下降[2-5],以上结果则表明HTPB 在UPR 链中的引入不仅提高了断裂伸长率,而且还使拉伸强度增大,其机理分析见3.4 节。
如图4 所示比较了不同HTPB 质量分数的UPR 在室温和低温下的拉伸模量。从图中可见,低温时各样品的拉伸模量均高于室温时的拉伸模量,符合一般高分子及其复合材料的特性。但显然,当w(HTPB)<4%时,UPR 的拉伸模量几乎并未因HTPB 的引入而降低。表明UPR 的刚性也几乎不受HTPB 引入的影响。
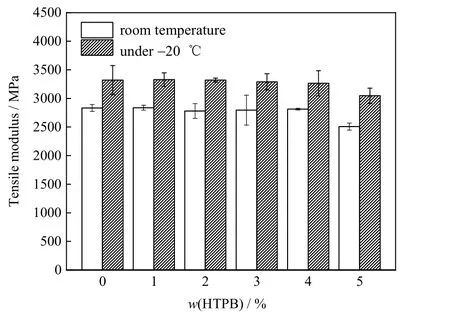
图4 不同HTPB 质量分数UPR 的室温与低温(-20 ℃)拉伸模量Fig.4 Tensile modulus of UPR with different HTPB contents at room temperature and -20 ℃
3.3 冲击性能
纯UPR 和UPR-HTPB 在常温及低温(-20 ℃)下的冲击性能如图5 所示。从图中可见,无论是室温或是低温条件下,随着HTPB 质量分数的增加,改性UPR 的冲击强度均呈上升趋势。当w(HTPB)=5% 时,改性UPR 常温和低温下的冲击强度分别达到(3.97±0.16)和(2.75±0.05) kJ·m-2,分别比纯UPR 增大了132%和167%。
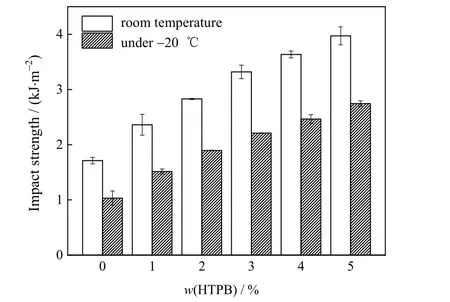
图5 不同HTPB 质量分数UPR 的室温与低温(-20 ℃)冲击性能Fig.5 Impact properties of UPR with different HTPB contents at room temperature and -20 ℃
3.4 机理分析
进一步通过扫描电镜(SEM)观察纯UPR 和不同质量分数HTPB 改性UPR 的冲击断面形貌,结果如图6 所示。由图6(a)可以看出,纯UPR 冲击断面上出现少量的大裂纹,断口平滑、裂纹扩展方向单一,呈现明显的脆性断裂的特征,表明耗散的冲击能小。当w(HTPB)=1% 时,如图6(b)所示,冲击断口表面的裂纹数量增加,裂纹在向前扩展时因预聚物链的柔性出现了偏转、分叉现象。裂纹的偏转增加了裂纹扩展的路程;裂纹尖端的分叉,减少了裂纹前进的动力,同时产生了更多的裂纹,起到了应力分散的作用,从而提高了冲击强度。但此时仍为脆性断裂。当w(HTPB)增加到2%~5% 时,如图6(c)、(d)、(e)、(f),断口表面形貌有了明显的区别,冲击断面变得粗糙,并出现了韧窝,产生的剪切屈服可吸收更多的冲击能,呈现出明显的韧性断裂的形貌。由图6(d)~(f)可见,随着HTPB 质量分数的增加,韧窝明显增多,使样品在断裂过程中更易产生局部的塑性变形,进一步提高冲击强度。在图6(c)~(f)中,断面上出现的小孔可能是HTPB 在固化过程中因与UPR 微相分离而产生的橡胶微粒,当受冲击时这种微粒通过诱发银纹也可吸收一部分能量。

图6 不同HTPB 质量分数的UPR 的冲击断面形貌Fig.6 SEM micrographs of fractured surface of UPR with different HTPB contents
通过在不饱和聚酯预聚物链中引入HTPB,从预聚物自身的柔性、预聚物固化后形成的韧窝和微相分离形成的橡胶相微粒等方面,分别赋予了UPR 的应力分散、剪切屈服和诱发银纹等能量吸收作用,因而有效提高了树脂的韧性。
采用传统方法制得的端基功能化液体橡胶,难以保证它们的双端基均含有反应性基团,现有的功能化液体橡胶改性UPR 的研究多采用共混-共固化的方法[2,4,7,41],即将传统的不饱和聚酯预聚物与端基功能化的液体橡胶在活性稀释剂中共混,通过固化反应将它们交联在一起。曾庆乐等[2]和葛曷一等[41]分别考察了不同活性端基的聚氨酯液体橡胶与UPR 共固化产物在室温下的力学性能,并分别用SEM 观察了冲击断面形貌。他们在实验过程中加入液体橡胶的质量分数均在7% 以上,冲击强度虽有提高,但拉伸强度、弹性模量均大幅度下降。如文献[41]报道,当液体橡胶质量分数为8% 时,断裂伸长率仅提高7.1%~52.9%,而拉伸强度下降最高达21% 以上。SEM 照片显示,纯UPR 的冲击断面上同样分布着许多细而直的河流线,而质量分数为10%的橡胶增韧的UPR 则分布着大小不等的2 种球形橡胶颗粒[41]。其中大颗粒(直径为10~30 μm)起阻止裂纹发展的作用,小颗粒(直径为0.1~1 μm)起剪切形变作用。这种大颗粒橡胶虽可吸收能量,提高树脂的冲击强度,但较大程度地降低了树脂的拉伸强度。液体橡胶分子仅在端基上与不饱和聚酯预聚物形成了交联网络或支化链(仅一端含有反应性基团时),较好地解决了橡胶颗粒在基体树脂中的均匀分散问题,但仍易产生使拉伸强度明显降低的过大橡胶颗粒。
Cherian 等[7]考察了端羟基聚丁二烯、环氧化天然橡胶、端羟基天然橡胶等功能化橡胶与传统UPR共固化产物在室温下的力学性能。发现拉伸强度、断裂伸长率、冲击强度均随橡胶质量分数的增大有一个先增高后降低的过程。对于端羟基聚丁二烯(摩尔质量为2 620 g·mol-1)改性的UPR 体系,当橡胶质量分数为2.5% 时,拉伸强度、断裂伸长率和冲击强度均达到最大值,分别比纯UPR 增大了61%、50% 和112%。文献[7]未解释这种同时增韧和增强的原因。如前所叙,UPR-HTPB 改性体系同样具有同时增韧和增强的特性,但各项力学性能先增大后减小的趋势并不明显;尤其在低温下,在实验所进行的0~5% 的HTPB 质量分数范围内,拉伸强度、断裂伸长率和冲击强度都随HTPB 质量分数的增加而增大。这一方面是因为将HTPB 无规地引入不饱和聚酯预聚物链中,使其固化时发生微相分离,不易形成较大粒径的橡胶颗粒。因此在提高树脂柔性的同时阻滞了拉伸强度和模量的下降。另一方面,与文献[41]所采用的聚氨酯液体橡胶不同,本文作者所采用的高顺式HTPB 内含有大量C=C 双键,固化过程中可与UPR 和活性稀释剂苯乙烯中的不饱和键发生反应,增大了交联密度,因而具有更高的机械强度。而在室温下,之所以会出现拉伸强度随HTPB 增多而增大的效果减弱(如图3(a)),是因为在UPR 链中引入过多的HTPB仍可能形成大尺寸的橡胶微团。
3.5 动态力学性能
图7 显示了不同HTPB 质量分数改性UPR 样品的动态力学性能。由图7(a)可见,所有样品均表现出明显的无定型聚合物的动态力学特征。纯UPR 在-100 ℃条件下的储能模量为4 969 MPa,且随温度变化较小;当温度上升至约50 ℃时,因为聚酯链的松弛,储能模量开始急剧下降,由玻璃态向橡胶态转变;当温度升至约150 ℃时,聚酯链几乎完全松弛,储能模量趋于稳定,为34 MPa。从图中可以看出,改性UPR 在-100℃时的储能模量均高于纯UPR,表明它们的刚性更强,而在150 ℃时则正好相反。可能因为,UPR 在-100 ℃时处于玻璃态,即使是HTPB 链段,因其诱导结晶所致的自增强效应[39]及大量双键带来的交联密度增大,使模量增大,刚性增强;而在150 ℃时,各样品均处于橡胶态,柔性长链HTPB 分子的引入使UPR 变软,模量下降。
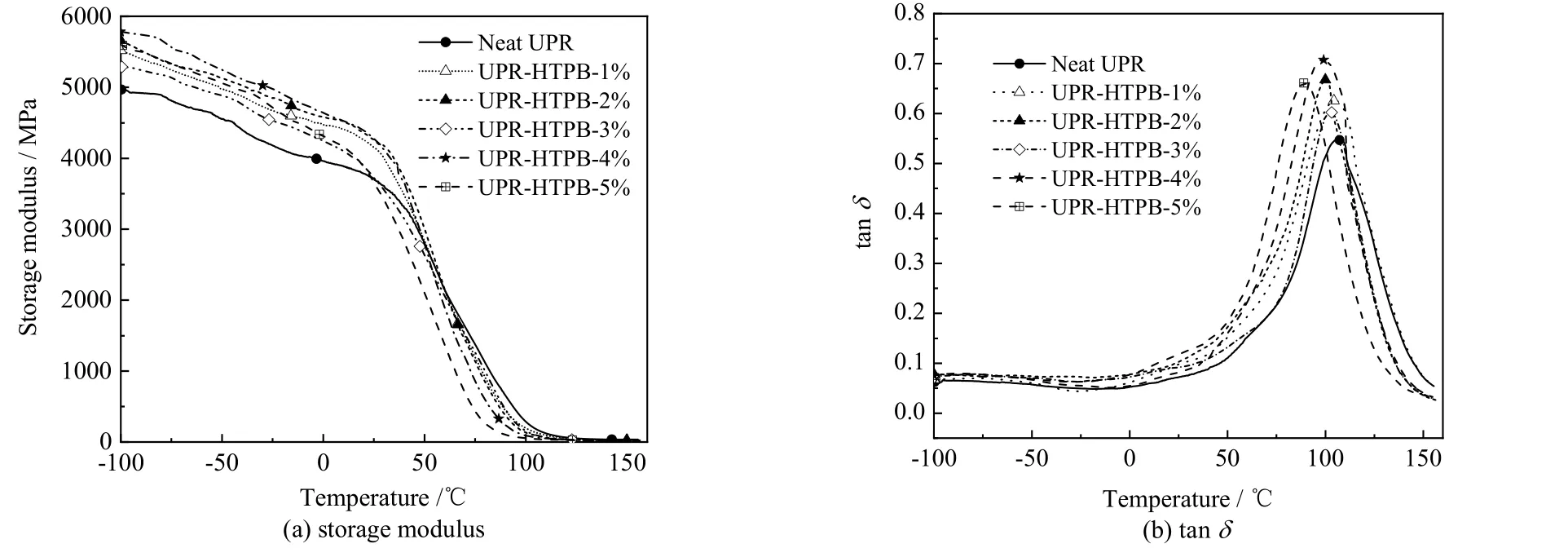
图7 不同HTPB 质量分数的UPR 的动态力学性能Fig.7 Dynamic mechanical properties of UPR with different HTPB contents
UPR-HTPB 损耗因子tanδ随温度的变化如图7(b)所示。从图中可见,各曲线均只有一个峰值,对应唯一的玻璃化转变温度,表明各固化UPR 样品的相分离并不明显。由图可见,随着HTPB 质量分数的增加,玻璃化转变温度下降;当w(HTPB)=5%时,玻璃化转变温度由108.1 ℃降至89.4 ℃,表明HTPB 的引入使UPR 柔性增加。
由图7(b)可见,各UPR-HTPB 的tanδ值均大于纯UPR。这是因为HTPB 的引入提高了UPR 内部高分子链段的运动能力,使分子更易发生相对位移,消耗更多的能量。
3.6 热稳定性
如图8 所示为纯UPR 和UPR-HTPB 的热失重(TGA)和微分热失重(DTG)曲线图。由图可见,各种UPR 样品的热分解过程均只有一个阶段,且初始分解温度十分相近,均在170 ℃左右;但初始阶段UPR-HTPB 的重量损失速度略低于纯UPR,且DTG 曲线峰值向高温方向略有移动,表明UPR- HTPB 具有更好的热稳定性。这与HTPB 的引入带来更高的碳-碳链比例有关。

图8 不同HTPB 质量分数的UPR 的热失重和微分热失重曲线Fig.8 TGA and DTG curves of UPR with different HTPB contents
4 结 论
以高顺式端羟基聚丁二烯液体橡胶为改性剂,合成了一系列不同HTPB 质量分数的不饱和聚酯预聚物,考察了它们固化后的力学性能和热性能。结果表明:
(1) 在常温或者低温(-20 ℃)下,不同HTPB 质量分数的UPR 均较纯UPR 有更高的断裂伸长率、拉伸强度和冲击强度,弹性模量则变化不大。室温下,冲击强度和断裂伸长率均随HTPB 质量分数的增加而增大,当HTPB 质量分数为5% 时,冲击强度和断裂伸长率分别比纯UPR 增大了132% 和124%;而拉伸强度则随HTPB 质量分数的增大先增加而后则略有下降,当HTPB 质量分数为3%时,拉伸强度最大,比纯UPR 增大了112%。低温下,冲击强度、断裂伸长率和拉伸强度均随HTPB 质量分数的增加而增大;当HTPB 质量分数为5% 时,冲击强度、断裂伸长率和拉伸强度分别比纯UPR 增大了167%、220%和186%,改性效果更为明显。
(2) 冲击断面形貌分析表明,HTPB 的加入改变了UPR 的微观结构,使其发生由脆性断裂向韧性断裂的转变。DMA 测试则显示,随着HTPB 质量分数的增大,UPR 的玻璃化转变温度明显下降;当HTPB质量分数为5%时,其玻璃化转变温度比纯UPR 降低了19 ℃。TGA 测定结果表明,含有HTPB 的UPR仍只有一个热分解阶段,且其较纯UPR 有更好的热稳定性。
(3) 综合分析各项实验结果, UPR 预聚物链中HTPB 的引入,可同时提高其固化产物的韧性和强度。这是因为HTPB 在不饱和聚酯预聚物链中的插入并参与交联反应,有效地阻止了弹性体在固化过程中的团块化聚集和大颗粒橡胶的形成,并增多了可显著吸收能量的微相分离区。
