儿童组织细胞坏死性淋巴结炎并发肝损害及血清铁蛋白升高1例并文献复习
2021-08-31唐学武甘李兰杨金潮王丽霞
唐学武,张 刚,甘李兰,杨金潮,王丽霞
(深圳市儿童医院,广东 深圳 518038)
组织细胞坏死性淋巴结炎(HNL)是一种罕见的以亚急性坏死性淋巴结肿大为特征的实体疾病,常与发热有关。在组织学上,该病的特点是皮质旁淋巴结扩张,有片状的、清晰的坏死区域,显示出丰富的核碎片、中性粒细胞和嗜酸性粒细胞的缺失。通过免疫组织化学(免疫组化)方法,该病组织中的组织细胞对髓过氧化物酶呈阳性反应,且T细胞明显占优势,余淋巴细胞少见。该病总体发病率低及症状、体征、辅助检查结果往往不典型,且目前临床医生和病理学家对这一疾病知之甚少,常导致误诊、漏诊,需要进一步研究。
1 临床资料
1.1一般资料 患儿,男,7岁零11个月,学龄期儿童,于2020年8月15日13:28以“间断发热1月余”收入本科。患者于1月余前无明显诱因出现发热,热峰39.3 ℃,发热间期约12 h,无畏寒、寒战,无抽搐,口服退热药后体温可降至正常,伴流清涕、打喷嚏,有颈部疼痛,伴有下肢瘙痒皮疹,无鼻塞,无咳嗽、咳痰,无喘息气短等不适,1个月前多次就诊于当地社康医院,考虑“上呼吸道感染”,给予“磷酸奥司他韦、六神丸、鱼腥草”口服3 d后,体温维持正常范围5 d。之后再次出现发热,热峰增至40+℃。发热间隔较前缩短,遂于深圳市龙岗区人民医院门诊就诊,完善检查示抗“0”增高,给予注射苄星青霉素静脉滴注1次,患儿仍发热,再次复诊于当地社康医院,行肝功能示肝酶升高,遂转入深圳市龙岗区人民医院住院治疗7 d,住院期间复查抗“0”示686 IU/mL,给予头孢呋辛4 d抗感染、护肝治疗,未再诉颈部疼痛,然后体温稳定2 d出院。1周前患儿再次出现发热,峰值40 ℃,伴双下肢可见针尖大小的红色斑丘疹,压之褪色,瘙痒,无关节疼痛,复诊于当地社康医院,给予阿奇霉素静脉滴注3 d后,患儿仍反复高热,今为进一步治疗来本院就诊,门诊以“发热查因”收入院。自发病以来,患儿精神状态一般,饮食、睡眠情况一般,大小便正常,近期体重无明显下降。
1.2入院后查体 体温:36.6 ℃,脉搏:96次/分,呼吸:18次/分,血压:106/64 mm Hg(1 mm Hg=0.133 kPa),血氧饱和度:100%(未吸氧下),神清,精神反应可,双下肢可见针尖大小的红色斑丘疹,压之褪色,无渗出,无脱屑。面色红润,咽充血,扁桃体Ⅱ度肿大,未见分泌物。左右颈部可触及多个肿大淋巴结,1.5 cm×1.5 cm,质地软,活动度可,轻度压痛。腹软,未触及包块,肋下触及肝大,肋下2 cm,质软,无压痛。心、腹、神经系统查体未见明显异常。
1.3入院前辅助检查 2020年7月30日深圳市龙岗区人民医院:EB病毒衣壳抗原、EB病毒抗体阴性。2020年8月2日深圳市龙岗区人民医院:抗“0”686 IU/mL;8月7日复查抗“0”672 IU/mL;EB病毒DNA阴性;肥达氏反应+外斐氏阴性;肺炎衣原体抗体二项阴性;肺炎支原体免疫球蛋白M(IgM)阴性;肺炎支原体IgG阳性;肝酶:谷草转氨酶(AST)118 U/L,谷丙转氨酶(ALT)92 U/L。2020年8月 11日深圳市龙岗区人民医院:血常规+C反应蛋白(CRP):白细胞3.74×109L-1,中性粒细胞绝对值1.95×109L-1,淋巴细胞绝对值1.54×109L-1,中性粒细胞比例52.1%,血红蛋白110 g/L,血小板357× 109L-1,超敏CRP 2.28 mg/L。2020年8月 14日本院门诊:血常规+CRP:白细胞3.31×109L-1,中性粒细胞绝对值1.87×109L-1,淋巴细胞绝对值1.30×109L-1,中性粒细胞比例56.4%,血红蛋白111 g/L,血小板319×109L-1,超敏CRP 2.62 mg/L;血涂片:中性杆状核0.02,中性分叶核0.47,淋巴0.46,单核0.05,白细胞形态、红细胞、血小板形态未见异常。
1.4入院后辅助检查 心脏标志物+基础代谢1+风湿+血脂+肝酶+铁蛋白(FER)+体液免疫+肝脏代谢:IgA 3.12 g/L、乳酸脱氢酶(LDH)796 U/、白蛋白34.7 g/L、肝酶:AST 29 IU/L,ALT 74 IU/L;电解质:钙201 mol/L、钠130.7 mol/L、铁蛋白2 236.94 ng/mL。CRP+ 血常规(五分类) :白细胞2.710×109L-1,淋巴细胞比例44.0%、中性粒细胞绝对值1.44×109L-1、单核细胞绝对值0.10×109L-1、 血小板310×109L-1、血红蛋白100 g/L、淋巴细胞绝对值1.2×109L-1,超敏CRP 1.25 mg/L;降钙素原0.15 ng/mL;红细胞沉降率(ESR) 38 m/h。乙型病毒性肝炎+人类免疫缺陷病毒(HIV)/丙型肝炎病毒(HCV)+TP无异常。结核免疫分析无异常。自身抗体+抗核抗体(ANA)+抗中性粒细胞胞浆抗体(ANCA)无异常。肿瘤标志物无异常。总IgE(化学发光法):524.80 IU/mL。吸入过敏源过筛(Phadiatop)试验+f×5+总IgE (定量):f×5鸡蛋白、牛奶、鱼、小麦、花生、大豆(酶免荧光法0.20 KIU/L;Phadiatop吸入物筛查84.10 KIU/L。复查CRP+血常规(五分类):白细胞5.51×109L-1、中性粒细胞绝对值2.55×109L-1、淋巴细胞绝对值2.39×109L-1、中性粒细胞比例46.2%、淋巴细胞比例43.4%、单核细胞比例10.2%、血红蛋白104 g/L、血小板283×109L-1、超敏CRP 3.49 mg/L。8月22日复查心脏标志物+基础代谢1+血脂+肝酶+FER+肝脏代谢:LDH 1 052 IU/L、清蛋白33.6 g/L、氯97.5 mmol/L、AST 275 IU/L、ALT 143 IU/L、钙2.15 mmol/L、钠125.4 mmol/L、铁蛋白4 158.99 ng/mL。后期复查,CRP+血常规:白细胞4.02×109L-1、中性粒细胞绝对值2.33×109L-1、淋巴细胞绝对值1.43×109L-1、中性粒细胞比例57.9%、淋巴细胞比例35.6%、血红蛋白103 g/L、血小板287×109L-1、超敏CRP 2.06 mg/L。肝功能:AST 136 IU/L、ALT 185 IU/L、铁蛋白1 476.03 ng/mL。血清总IgE 588.3 IU/mL。
1.5病原学 呼吸道病原体聚合酶链式反应13项:呼吸道合包病毒阳性;EB病毒DNA (血):阴性;咽拭子EB病毒DNA测定(定量):(X)<4.00E+2 copies/mL;血培养无异常。
1.6影像学 头颅+鼻窦CT:头颅CT平扫未见异常,双侧下鼻甲肥大。胸部CT平扫未见异常,请结合临床。心脏超声无异常。腹部超声:肝脏弥漫性增大,肝门淋巴结增大,双侧颈部淋巴结增大。
1.7病理学检查 骨髓检查:全片见少量组织细胞,偶见噬血组织细胞。颈部淋巴结活检(图1):淋巴结结构部分保存,副皮质区扩大,散在不规则片状碎眉样坏死,坏死灶内及其边缘处组织细胞增生,坏死区边缘免疫母细胞等转化性淋巴细胞增生,(左颈部)考虑HNL(Kikuchi淋巴结炎)。全自动免疫组化结果(A1):ALK(-),CD123(散在或灶状+),CD15 (粒细胞+),CD3(局灶T细胞+),CD30(个别细胞+),CD20(残存的滤泡区见+),CD34(内皮+),CD38 (浆细胞+),CD4 (T细胞+),CD45(+),CD68 (组织细胞+),CD8 (T细胞+),Pax -5 (残存的滤泡区见+),S-100(散在+)。特殊染色结果(A1):过碘酸-希夫染色(未见真菌菌丝及孢子),抗酸染色(-)。分子病理结果(AI):EB病毒原位杂交EBERs(-)。
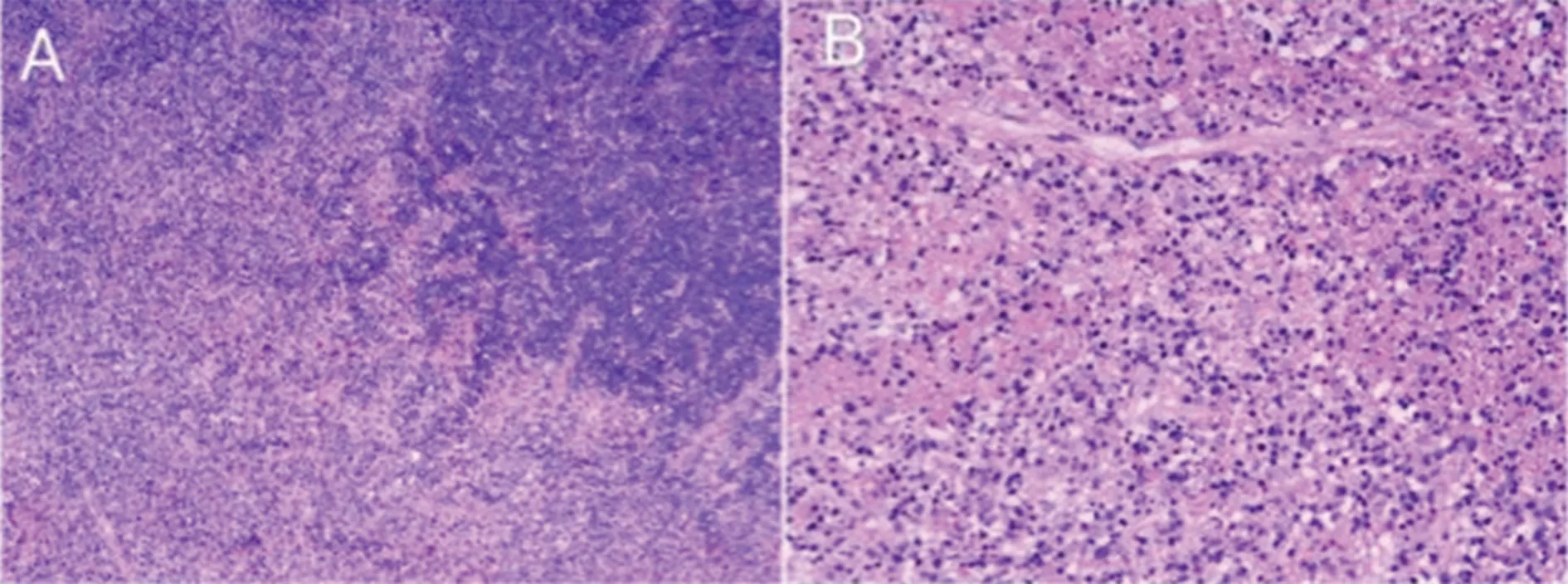
A.淋巴结结构部分保存,副皮质区扩大,散在不规则片状碎眉样坏死,坏死灶内及其边缘处组织细胞增生,坏死区边缘免疫母细胞等转化性淋巴细胞增生(免疫组化,200×);B.全自动免疫组化结果(A1):ALK(-),CD123(散在或灶状+),CD15 (粒细胞+),CD3(局灶T细胞+),CD30(个别细胞+),CD20(残存的滤泡区见+),CD34(内皮+),CD38 (浆细胞+),CD4 (T细胞+),CD45(+),CD68 (组织细胞+),CD8 (T细胞+),Pax -5 (残存的滤泡区见+),S-100(散在+)(免疫组化,40×)。
1.8诊疗经过 入院后予以阿莫西林舒巴坦静脉滴注抗感染(8月17—23日),苯联双酯片、葡醛内酯片护肝治疗,患儿每天反复发热,热波39.0~40.5 ℃,但发热间隔时间延长。8月20日行淋巴结活检: 考虑HNL(左颈部),8月24日开始给予泼尼松每次10 mg,每天1次[0.6 mg/(kd·d)]口服治疗;8月25日体温恢复正常,之后未再发热,双下肢皮疹逐渐减退,颈部淋巴结逐渐缩小,无压痛,肝脏无肿大,肝功能中AST、ALT值、血清铁蛋白值逐渐降低。遵医嘱泼尼松每半月减量4 mg,出院回家后继续口服泼尼松3个月,定期随访。
2 讨 论
HNL最早分别由日本的Kikuchi和Fujimoto提出,故又名Kikuchi病或Kikuchi-Fujimoto病(KFD)[1],是一种累及淋巴结的全身性、良性、自限性罕见的起源不明的淋巴结炎[2]。由于该病较为罕见,并且可以累及多种组织器官,个体临床表现差异性大,目前该病发病因素及机制尚不明确。
2.1病因和发病机制 该病病因至今尚不明确,目前考虑主要与病毒感染和自身免疫紊乱有关。该病多发生在亚洲国家,西方国家少见,且儿童少见。众多研究报道,病毒感染和自身抗原的交叉反应可以是该病的始动病因,但该病原因与病原体阳性目前没有明确,故病毒感染作为HNL的病因仍存在一定争议[3- 4]。国内研究显示,从32例病原学检查中显示EB病毒、支原体、巨细胞病毒、柯萨奇病毒、腺病毒、单孢病毒、流感病毒,还有病原菌混合感染[5-6]。有科学家认为,该病的发病机制为有一定遗传倾向的机体在被病毒感染、地域(亚洲国家为主)、环境等多种因素的强烈刺激下,TH淋巴细胞被过度激活,进一步引发淋巴细胞介导过度免疫应答反应[7]。本病例中呼吸道病原体13项中呼吸道合包病毒阳性,总IgE水平升高10倍,且TH1/TH2细胞因子可见白细胞介素-4(IL-4)、IL-10、干扰素-γ(INF-γ)水平升高,考虑该病不仅与病原体有关,可能还存在自身免疫紊乱情况。
2.2组织细胞病理学情况 淋巴结活检是该病最终确诊手段,病理学显示可见淋巴结正常结构消失,副皮质附近有大片坏死,周围有大量组织细胞而无粒细胞浸润,周围有大量CD68/髓过氧化物酶(MPO)组织细胞,CD68/CD123浆细胞样树突状细胞,少数小到大尺寸的CD8淋巴细胞和免疫母细胞。国外研究显示,在组织学上,HNL的特点是皮质旁淋巴结扩张,有片状的、清晰的坏死区域,显示出丰富的核碎片和中性粒细胞、嗜酸性粒细胞的缺失。病变中T细胞明显占优势(主要为CD8阳性细胞),B细胞很少[8]。CUGLIEVAN等[9]为探讨凋亡细胞的细胞类型,对12例HNL进行了细胞毒性T细胞或NK细胞的细胞毒性颗粒T细胞内抗原(TIA-1)免疫组化染色,1/4~1/2以上的凋亡细胞对TIA-1呈阳性,一些核碎片也呈阳性,还发现HNL的坏死性病变由核碎片、凋亡细胞、组织细胞和淋巴细胞组成。淋巴细胞主要为CD8+T细胞或CD4+细胞,而B细胞和NK细胞很少观察到。TIA-1阳性淋巴细胞数量与CD8阳性细胞数量的关系比CD4细胞数量的关系更为密切。在双染色中,TIA-1阳性淋巴细胞主要为CD8阳性,但很少有CD4阳性。在该病中,CD8阳性的细胞毒性T细胞可能发生凋亡[9]。在32例HNL患儿的淋巴结活检显示中,大体所见为灰白色样组织,光镜所见为淋巴结副皮质区出现淡染、不规则形状的片状病变,同时可见明显的核碎裂或凝固性坏死,其内可见组织细胞、浆样树突细胞及混杂的淋巴样细胞,没有或很少见到中性粒细胞,且无苏木精小体;免疫组化染色显示组织细胞标记CD68(+)、CD163(+),另可见中等量T细胞(CD3),而B细胞(CD20)很少[5]。本病例淋巴结活检显示,淋巴结结构部分保存,副皮质区扩大,散在不规则片状碎眉样坏死,坏死灶内及其边缘处组织细胞增生,坏死区边缘免疫母细胞等转化性淋巴细胞增生,与上述研究的淋巴病理学检查保持大同小异。
2.3临床表现、治疗和预防复发 该病主要临床表现是反复发热和淋巴结肿大,发热往往是突出临床表现或者首发症状,1/3以上的患者为不明原因的长期反复发热,早期为低热,后期以高热为主,热型无规律,可持续好几周,甚至数月;淋巴结肿大主要是颈部的颈后三角区淋巴结肿大,疼痛,通常发生于一侧,全身广泛性浅表淋巴结肿大比较罕见。临床表现还可伴有恶心、呕吐、体重减轻、头痛和关节痛,这些临床表现往往缺乏特异性,还有比较罕见的结外延伸,包括皮肤、眼睛和骨髓定位,皮肤表现为全身散在斑点状皮疹。有研究显示,皮疹活检病理表现与淋巴结组织细胞病理保持一致[10-11]。该病主要依赖于淋巴结活检来确诊。有研究探讨HNL可受累多个器官情况,34例患者均累及淋巴结,还累及其他系统或器官累及,比如血液者占67.6%,皮肤者占23.5%,肺脏占14.7%,浆膜腔者占11.8%,脑膜及关节占比重极少[12]。纵多研究显示,该病累及器官可导致各种并发症,如皮肤(过敏性皮疹)[10]、肺部(肺炎甚至肺出血)[13]、神经系统(无菌性脑膜炎、小脑共济失调)[14]、心脏(无菌性心肌炎、心包炎)[15],以及风湿免疫性疾病[15],而多器官受累者病情严重者甚至引起多器官衰竭而死亡[16]。在本病例中,合并肺部感染、皮肤疾病,还引起肝脏肿大及损害,且肝功能异常(AST、ALT值升高好几倍),血清铁蛋白水平明显升高,目前国内外研究中该病累及肝脏肿大及损害和血清铁蛋白明显升高报道甚少。在一些炎症或恶性病变中,如肝癌、肺癌、胰腺癌、白血病、霍奇金病、多发性骨髓瘤等恶性肿瘤细胞可以合成和分泌铁蛋白,铁蛋白测定已成为恶性肿瘤辅助诊断指标之一,并且急、慢性肝炎或其他肝病时血清铁蛋白也明显增高。该病引发血清铁蛋白明显升高和肝功能异常(AST、ALT值明显升高),考虑可能患儿肝功能异常引发的血清铁蛋白明显升高,同时也要警惕后期恶性肿瘤的演变。
目前认为激素治疗是治疗该病的主要手段,且预后良好[17-18],但合理使用激素来治疗该病至关重要。有研究显示,多器官受累组(累及3个及以上器官)激素治疗者初始剂量比非多器官受累组(2个或1个)大,且多器官受累组激素治疗时间也比非多器官受累组长[12]。本病例在2020年8月24日给予泼尼松每次10 mg,0.6 mg/(kd·d)每天1次口服治疗,并遵医嘱逐渐每半月减量4 mg,患儿出院口服激素3个月。患儿自服用泼尼松后的第2天就未再发热,双下肢皮疹明显减退,无瘙痒,颈部淋巴结逐渐缩小,无压痛,但仍肝脏肿大,肝功能中AST、ALT值仍比正常值高3倍,且血清铁蛋白从4 158.99 nm/mL降至2 236.94 nm/mL,再复查为1 476.03 nm/mL;加用激素治疗期间,患儿血清总IgE(524.8 IU/mL),复查存在一次升高(588.3 IU/mL),后期患儿未再发热,复查血总IgE逐渐恢复正常。有研究报道,1例HNL合并高IgE血症者通过使用激素,患者病情逐渐控制,血清总IgE逐渐下降,随后恢复正常,可能的主要原因是患者体内TH1/TH2细胞失衡,促使TH0细胞向TH2细胞增殖,进一步促进B细胞大量分泌IgE[19]。患儿现已出院,目前未再出现发热,淋巴结逐渐缩小,无压痛,无皮疹,肝功能和血清铁蛋白正常。该病有一定复发倾向,有研究发现无多器官受累者复发少见,多器官受累者复发率可能增高[20],应加强后期随访,注意避免感染,合理使用激素,预防复发。
综上所述,虽然目前认为HNL预后良好,且确诊该病尤为重要,但其发病因素及机制尚不清楚,而且关于其严重程度、器官受累情况及激素合理治疗的国内外相关研究报道较少,需要进一步研究。
