Ir(100)面上HAN催化分解反应机理
2021-08-30王海丰黄永民
胡 旭,刘 川,王海丰,黄永民
(1.华东理工大学 化学与分子工程学院,上海 200237; 2.上海空间推进研究所,上海 201112)
0 引言
单组元液体火箭发动机,是一种以单一的推进剂组元作为工质的发动机。相较于传统的双组元发动机,该发动机具有结构简单、可靠性高、推力控制精度高、成本低等特点,可应用于卫星、飞船、空间探测器、导弹弹头、运载火箭等飞行器的姿态控制动力系统。硝酸羟胺(HAN)基推进剂作为一种新型含能离子液体推进剂,通常是由HAN、燃料以及水等多种物质复配形成的氧燃共存体系,与传统肼类推进剂相比,该推进剂以其优异的能量特性、较低的冰点温度以及良好的贮存安全性等特点,被认为是新一代单组元液体火箭发动机的一种理想燃料。
HAN基单组元液体火箭发动机的工作过程中,推进剂首先通过燃料供给装置进入催化床,并在床内与催化剂颗粒接触,发生催化分解与燃烧现象,产生高温气体,最终通过喷管排出产生推力。因此,催化剂的筛选与制备以及HAN基推进剂在催化剂表面的分解燃烧过程将直接影响发动机的比冲性能和工作寿命。为促进HAN基推进剂的应用,国内外学者在该领域进行了一定的研究工作。
在催化剂的筛选与制备研究过程中,分别研究了铱、铂、铼等贵金属对HAN的催化效应,研究结果表明铱基催化剂能有效促进HAN基单元推进剂的分解。对于HAN的催化分解反应机理及动力学,Chang基于稳态假设,提出了一套HAN与燃料相互反应作用的简化动力学模型,并通过理论计算获得了亚硝酸(HNO)和羟胺离子(NHOH)的产成速率。研究结果表明,HNO是HAN催化分解反应的重要产物,其产成速率会对HAN基推进剂的分解与燃烧特性产生显著的影响。当提高燃料的浓度或增加氧化反应的速率时,HNO的生成速率会呈现下降趋势。吴珊珊等采用气质联用仪对HAN基液体推进剂的催化分解产物进行分析,检测发现其催化分解产物主要为N、NO、CO、NO和HO。文献[17—18]针对液体推进剂的相变过程,首先提出了基于催化反应实验的一级反应催化相变模型,进而基于高温热解实验数据提出了分段的相变模型,最终拟合获得HAN基推进剂的宏观分解反应速率表达式。
综上所述,对于HAN基液体推进剂的催化分解燃烧过程主要以实验为主,对于其催化机理的相关研究较少。由于HAN分解产物的种类众多,且部分氧化性产物(NO、NO等)会与推进剂内其余组分发生相应的化学反应,进而影响推进剂的催化分解过程。虽然通过实验方法可以获得相应的规律及产物分布,但其所用的催化剂都为贵金属,实验成本较高,且对于微观结构难以精确控制。随着量子化学计算方法和计算机技术的发展,可采用量子化学算法获得相应的催化分解燃烧微观机制,获得分解反应进程中的产物分布。另外在一些理论研究中,大多针对单个HAN分子的催化分解机理进行研究,而在真实发动机启动过程中,HAN基推进剂是以液态的形式与催化剂接触并开始发生分解反应。因此本文将以HAN双分子缔合结构模拟液态HAN中分子间作用力的影响,利用量子化学计算方法,研究HAN双分子缔合结构在Ir表面上的初始催化反应机理,获得HAN缔合结构的初始分解反应路径及能垒。为HAN基推进剂和催化剂体系的研制以及HAN基单组元液体火箭发动机燃烧室设计提供理论依据。
1 计算方法
利用从头算算法(Ab
Initio
)中的组合方法Gaussian-4(G4)对硝酸羟胺及其双分子缔合形态进行结构和能量进行计算。该方法是建立在ab initio分子轨道理论的基础之上,计算中的基本能量是通过MP4/6—31G(d
)后自洽场计算获得的。其次,以基本能量的基准,采用较低级别的Ab
Initio
计算方法进行外推计算,在计算过程中通过加入一系列的能量修正值,来模拟较大基组下CCSD(T)方法的能量计算值。通过G4方法计算获得的各分子能量的平均误差可控制在0.83 kcal/mol以内。通过G4方法进行计算可以准确获得相应分子的结构及热力学参数(焓、熵、吉布斯自由能等)。计算过程由Gaussian09程序完成。对于催化剂结构及相应催化反应机理的计算,应用具有广义梯度近似(GGA)的 Perdew-Burke-Ernzerhof (PBE)函数来计算交换相关能量,利用该方法计算获得的常见分子的原子化能与实验值的平均误差小于8%,能准确预示反应中的分子结构与能量。电子—离子间的相互作用是利用缀加平面波基组(PAW)展开方法,平面波基组的截断能取值为450 eV。SCF自洽迭代的收敛精度为1.0×10eV。对于表面吸附及催化分解反应的模拟过程中,催化剂表面由一个(4×4)的单元,计算模型为3层结构,最底层为固定结构,对催化剂的上面2层以及HAN分子结构进行优化计算,真空层的取值为12 Å,此时k
格点选取2×2×1。计算过程中采用的收敛准则为应力偏差小于0.05 eV/Å。对于反应路径中的过渡态结构通过微动弹性带(NEB)方法进行搜索,该方法主要是利用确定的反应物和产物结构,控制由反应物向产物发展路径中关键结构变化参数,并使剩余体系下能量达到最小值,过渡态结构则为反应路径上能量极大值点处的结构。上述计算将由VASP程序完成。2 结果与讨论
在计算过程中首先通过G4方法探究不同构象下的HAN分子结构及热力学参数,通过比对焓值与吉布斯自由能,确定稳定的HAN分子结构,并以此为基础,确认最为稳定的双HAN双分子缔合结构。利用GGA-PBE方法对Ir的晶格进行优化,并对Ir的3种晶面能量进行计算,选取合适的晶面进行催化分解反应路径的计算。
在反应路径的计算主要是通过GGA-PBE方法探究稳定稳定的HAN双分子缔合结构在所取晶面上的稳定吸附结构,通过判断吸附能筛选合适的吸附结构。针对可能存在的吸附结构,探究并优化计算获得解离吸附结构,并最终利用NEB方法搜索反应过渡态,构建反应势能面,获得反应能垒参数。
2.1 HAN结构优化
通过量子化学计算方法G4对HAN的结构和热力学参数进行计算,获得HAN可能存在的稳定构象及相应的热力学参数。计算结果表明,HAN主要存在有4种稳定的构象,各种构象结构如图1所示。
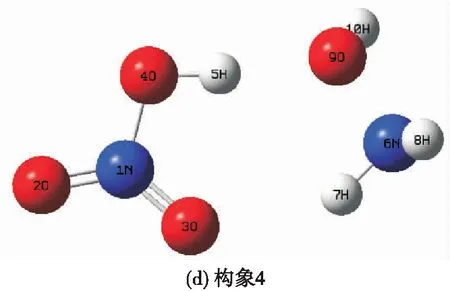
图1 HAN分子主要存在的4种构象Fig.1 Four main conformations of HAN molecular
其中,构象1和构象3是通过单个位点的氢键相互作用而结合,构象1为NHOH中的N与HNO中的OH形成氢键,O(4)—H(5)键长为1.018 Å,N(6)—H(5)键长为1.689 Å,O(4)—H(5)—N(6)的键角为173.578°;构象3为NHOH中的O与HNO中的OH形成氢键,O(4)—H(5)键长为1.001 Å,O(7)—H(5)键长为1.682 Å,O(4)—H(5)—O(7)的键角为171.971°。
构象2和构象4则是通过双位点的氢键相互作用而结合。在构象2中两个氢键分别为NHOH中的N与HNO中的OH以及NHOH中的OH与HNO中的O(3),其中N与OH所形成的氢键键长和键角分别为1.034 Å、1.625 Å和178.706°,O(3)与OH所形成的氢键键长和键角分别为1.902 Å、0.973 Å和157.38°;而在构象4中所产生的氢键分别为NHOH中的NH基团与HNO中的O(3)以及NHOH中的O(9)与HNO中的OH,其中NH与O(3)所形成的氢键键长和键角分别为2.182 Å、1.021 Å和146.792°,O(9)与OH所形成的氢键键长和键角分别为1.001 Å、1.677 Å和173.418°。
H
)和吉布斯自由能(ΔG
)可知,构象2的能量最低,其结构最为稳定。对于HAN而言,NHOH中的NH基团与HNO中的OH基团所形成的氢键更为稳定,而双氢键相互作用的结构比单氢键相互作用的结构稳定。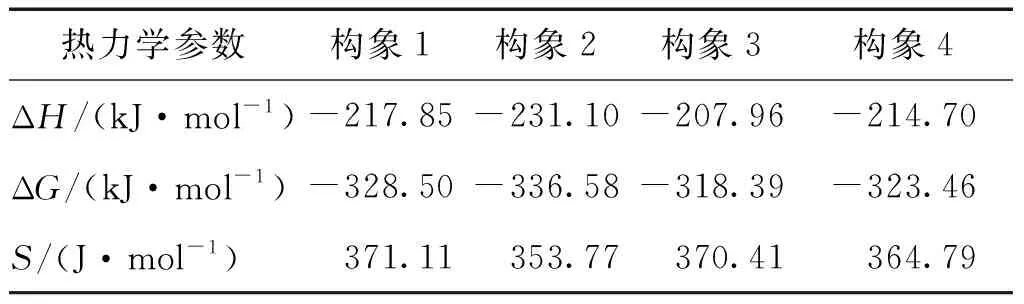
表1 标准状态和298.15 K温度下4种 构象HAN的热力学参数
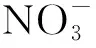
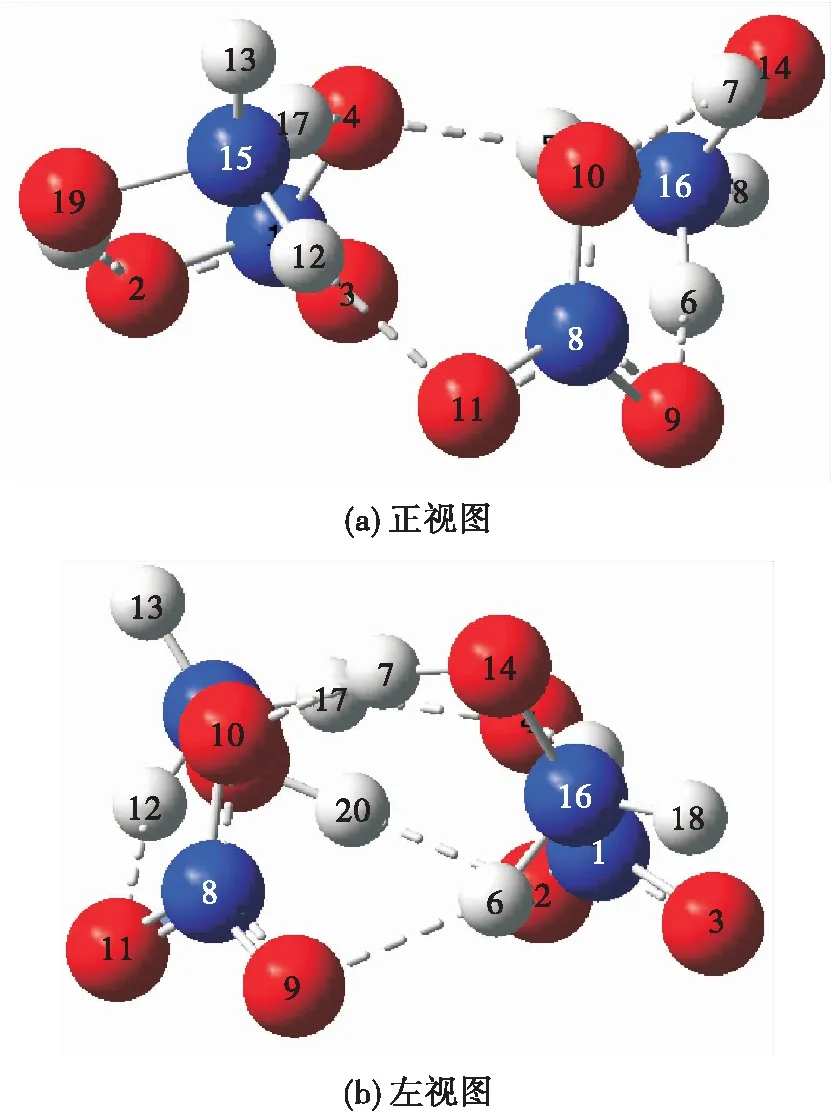
图2 HAN双分子缔合结构Fig.2 HAN bimolecular association structure
2.2 化学吸附计算
通过对Ir晶格进行优化计算,其主要为面心立方结构,其晶格边长为3.873 Å,主要可形成Ir(100)、Ir(110)和Ir(111)3种晶面结构,其表面能量分别为-16.22 eV、-17.51 eV和-4.63 eV。对比3种晶面能量可知,Ir表面最为稳定的晶面为Ir(100)和Ir(110)2种。本文将对HAN双分子缔合结构在Ir(100)面上的初始分解机理进行研究。
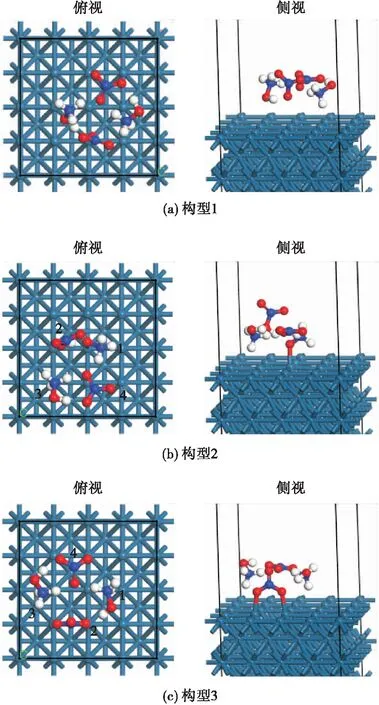
图3 HAN双分子缔合结构在Ir(100)面上的吸附结构Fig.3 Adsorption structure of HAN bimolecular association structure on Ir (100) surface
通过对获得的吸附构型下的吸附能进行计算,以此来判断不同吸附构型的稳定性,吸附能的计算公式为
E
=E
-E
-E

2.3 催化分解反应路径
根据2.2节中计算获得的稳定吸附结构,对HAN双分子缔合结构在Ir(100)面上的催化分解反应机理开展研究,针对构型2和构型3两种吸附结构进行计算,获得的了两种结构催化分解的过渡态结构和解离吸附构型,如图4和图5所示。
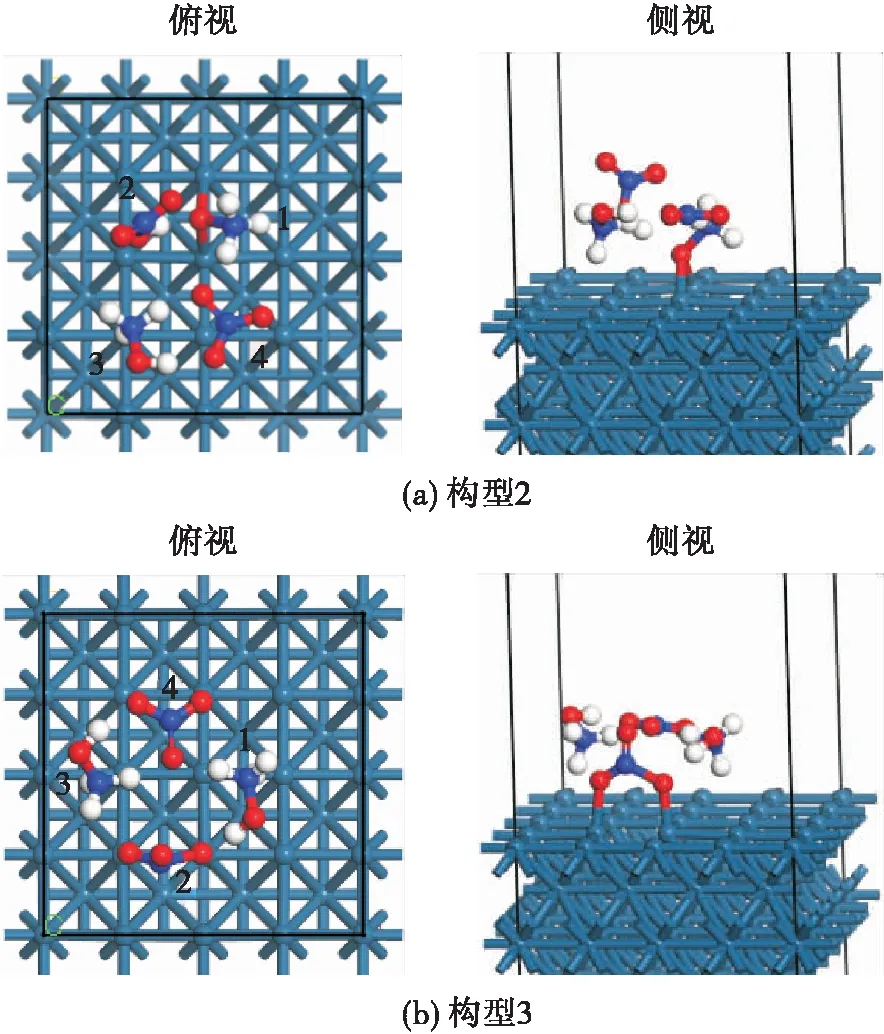
图4 HAN双分子缔合结构在Ir(100)面上的催化分解 反应过渡态结构Fig.4 Transition state structure of the HAN bimolecular association structure on Ir (100) surface in the catalytic decomposition reaction
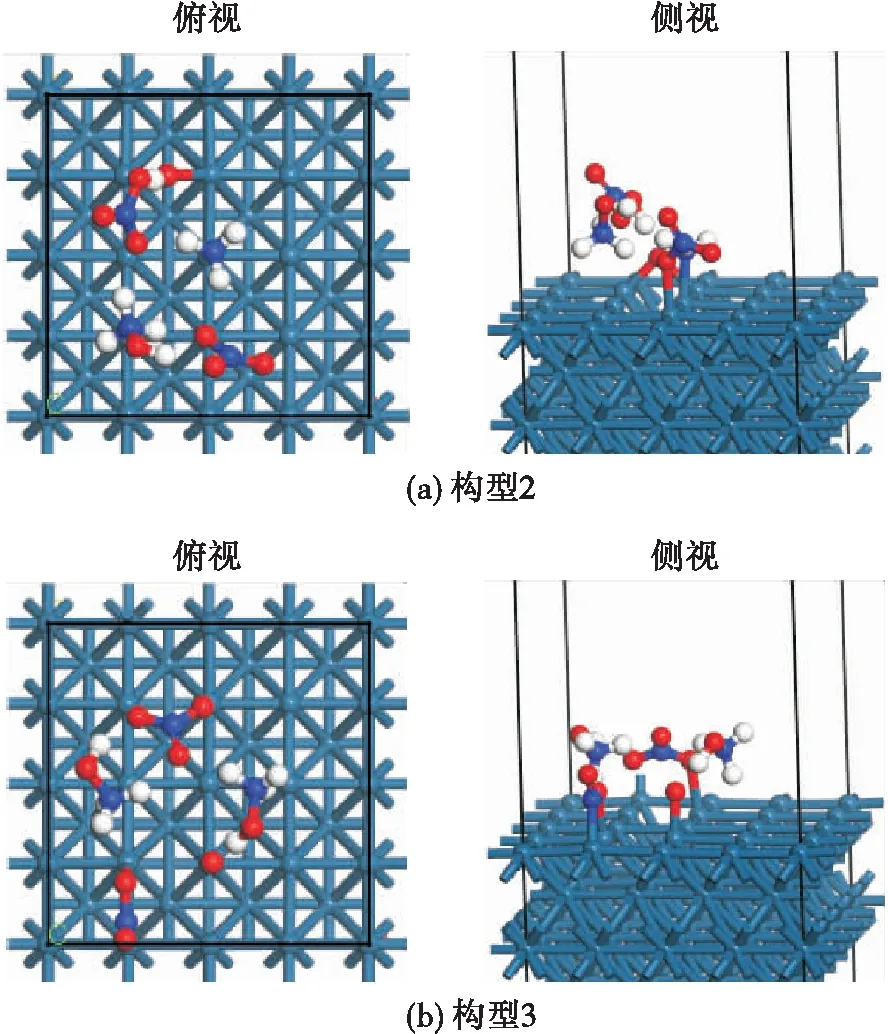
图5 HAN双分子缔合结构在Ir(100)面上的解离吸附结构Fig.5 Dissociation adsorption structure of HAN bimolecular association structure on Ir (100) surface
对比过渡态结构与反应物结构可知,在构型2中,其分解的初始路径主要是与Ir发生键合的NHOH基团(基团1)中O—N键的断裂,在其分解反应过渡态结构下,O—N键的键长与吸附结构相比出现小幅增加,由吸附结构下的1.43 Å增加至1.68 Å。
根据反应过渡态的分子结构,可以确定HAN双分子缔合结构在Ir(100)面上的催化分解反应的能垒及反应热,如图6所示。

图6 HAN双分子缔合结构催化分解能垒(kcal/mol)Fig.6 Energy barrier catalytic decomposition of HAN bimolecular association (kcal/mol)
由图可知,HAN双分子缔合结构的催化分解主要为放热反应,由于反应过程中均为N—O键发生断裂而出现分解,因此2个反应的能垒大体相当,对于NHOH的催化分解(构型2反应路径),其反应能垒较高,为12.68 kcal/mol。而对于构型3的分解,其反应能垒相对较低,为11.30 kcal/mol。由于2个反应的能垒相近,因此这两条反应路径在催化分解反应过程中会同时发生。
3 结论
采用第一性原理计算方法研究了HAN双分子缔合结构在Ir(100)面上的催化分解反应机理,计算结果表明:
1)在Ir(100)面上,HAN双分子缔合结构主要存在有3种吸附结构,其中最为稳定的结构主要为分子中的O与Ir的键合作用,其吸附能分别为-1.64 eV和-2.15 eV。
2)HAN双分子缔合结构在Ir(100)面上的分解反应过程存在有分解先后顺序,不含有双氢键的HAN分子会率先发生分解反应。对于羟胺吸附构型,其分解产物主要为NH、OH和NO,而对于硝酸分解主要产物为NHOH、NO和O。
3)HAN双分子缔合结构的催化分解均为放热反应,主要为体系中O—N键的断裂,NHOH和O—N键断裂所需能垒较高,反应释放能量高。2个构型下的催化分解能垒分别为12.68 kcal/mol和11.30 kcal/mol,HAN双分子缔合结构在Ir(100)面上的初始催化分解可能同时出现羟胺的分解与硝酸的分解。
通过相关研究,可以获得发动机起动阶段推进剂的初始反应机理及反应能垒,可用于探究推进剂与催化剂体系的极限反应条件,为HAN基推进剂和催化剂体系的研制以及HAN基单组元液体火箭发动机燃烧室设计提供理论依据。
