紫外-可见分光光度法间接测定西咪替丁的含量
2021-08-29何艳虹
林 瑜,丘 琴,2∗,何艳虹
(1.广西中医药大学 药学院,南宁 530200;2.广西优势中成药与民族药开发工程技术研究中心,南宁 530200)
西咪替丁是可选择性H2受体拮抗剂,用于消化性溃疡的治疗。目前,国内外检测西咪替丁的方法主要有高效液相色谱法(HPLC)[1-4]、液相色谱-质谱法(LC-MS)[5-8]、光谱法[9-13]、化学发光法[14-16]、核磁共振法(NMR)[17]、电化学法[18]、电位滴定法[19]等。现有的分析方法各有其优缺点,如HPLC与LC-MS的准确度和灵敏度较高,但仪器价格昂贵,操作复杂;化学发光法灵敏度高,但准确度和精密度较低;电位滴定法准确度好,但分析时间较长;NMR 能提供原子水平上的信息,但灵敏度相对较低。
西咪替丁易溶于甲醇、稀盐酸,可溶于乙醇,略溶于异丙醇,微溶于水,在218 nm 处有特征紫外吸收峰[20],而大多溶剂在210 nm 附近存在末端吸收,若直接采用紫外-可见分光光度法(UV-Vis),会对218 nm 处西咪替丁的测定造成干扰。
西咪替丁分子支链中的两个氨基氮原子均含有孤对电子,能与Cu2+发生络合,形成稳定的四元环配合物,结构如图1所示。此四元环配合物的紫外吸收波长红移至325 nm 处,从而避免了大多溶剂在210 nm 附近紫外吸收的干扰。据此,本工作建立了一种UV-Vis间接测定西咪替丁含量的方法,并成功应用于实际样品的测定。
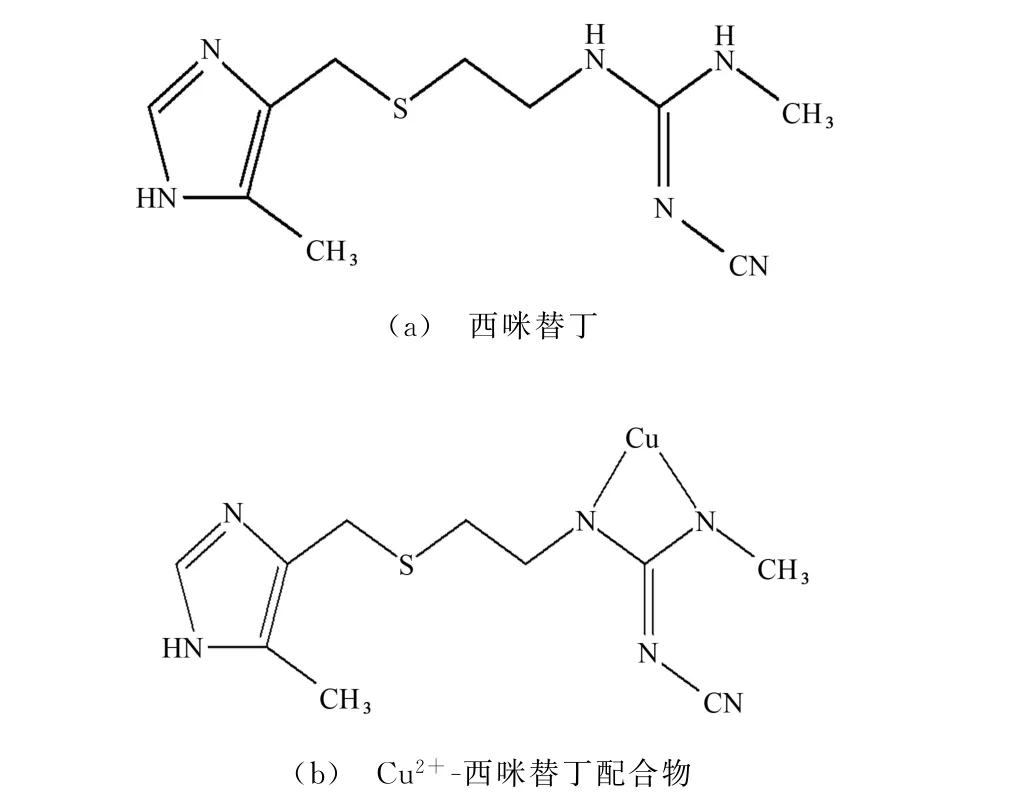
图1 结构式Fig.1 Structural formula
1 试验部分
1.1 仪器与试剂
UV-1780型紫外-可见分光光度计。
西咪替丁标准储备溶液:256.0 mg·L-1,称取0.025 6 g西咪替丁标准品于烧杯中,用0.1 mol·L-1盐酸溶液溶解并定容至100 mL 容量瓶中。使用时用0.1 mol·L-1盐酸溶液稀释至所需质量浓度。
Cu2+储备溶液:2.50 g·L-1,称取0.625 2 g CuSO4·5 H2O 于烧杯中,用水溶解并定容至250 mL容量瓶中。
Cu2+溶液:250 mg·L-1,移取2.50 g·L-1Cu2+储备溶液,用水稀释10倍制得。
pH 7的乙酸-乙酸钠缓冲液:称取25 g乙酸钠,用水溶解,加入0.1 mL 冰乙酸,混匀。将混合溶液转移至250 mL容量瓶中,用水定容。
西咪替丁标准品,纯度99.8%;西咪替丁片1,规格0.2 g·片-1;西咪替丁片2,规格0.2 g·片-1;西咪替丁片3,规格0.1 g·片-1。
所用试剂均为分析纯;试验用水为蒸馏水。
1.2 仪器工作条件
波长扫描范围为200~600 nm;光谱带宽为0.2 nm;分析波长为325 nm。
1.3 试验方法
将西咪替丁片剂1~3各20片研碎,分别称取样品粉末0.025 2 g,用0.1 mol·L-1盐酸溶液溶解并定容至100 mL 容量瓶中,摇匀,过滤后取滤液,即得西咪替丁样品溶液。取一定量的西咪替丁样品溶液、2 mL的250 mg·L-1Cu2+溶液于10 mL比色管中,用0.1 mol·L-1氢氧化钠溶液和0.1 mol·L-1盐酸溶液调节pH 至7 左右,再加入1.00 mL pH 7的乙酸-乙酸钠缓冲液,用水稀释至刻度,摇匀,于室温反应10 min,以空白试剂作参比,按仪器工作条件测定。
2 结果与讨论
2.1 金属离子的选择
固定西咪替丁标准溶液的量不变,分别加入等量的Cu2+、Ca2+、Al3+、Fe2+、Cd2+、Ni2+,摇匀后,用紫外-可见分光光度计对其进行全波长扫描,考察了西咪替丁在不同金属离子存在下的紫外-可见吸收光谱。结果显示,Cu2+与西咪替丁反应,可在325 nm 处产生新的最大特征紫外吸收峰,而Ca2+、Al3+、Fe2+、Cd2+、Ni2+与西咪替丁反应未产生明显紫外吸收峰,故试验选择Cu2+作进一步研究。
2.2 分析波长的选择
以水为参比,按仪器工作条件对不同溶液进行测定,所得紫外-可见吸收光谱图见图2。
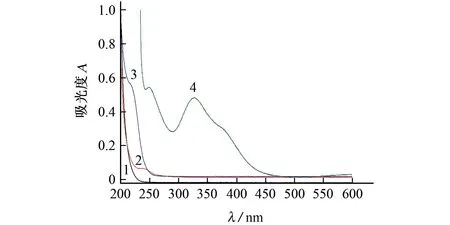
图2 不同溶液的紫外-可见吸收光谱图Fig.2 UV-Visible absorption spectrum of different solutions
由图2可知:西咪替丁在218 nm 处有较强的特征紫外吸收(曲线3),但此处容易受到溶剂末端吸收的干扰(曲线1),影响测定的准确度;曲线4在325 nm 处有新的特征紫外吸收,而曲线1~3在此处并没有特征紫外吸收,这是由于西咪替丁与Cu2+形成了配合物,从而产生了新的紫外吸收峰,故试验选择325 nm 作为分析波长。
2.3 溶液酸度的选择
移取4.00 mL的256.0 mg·L-1西咪替丁标准溶液、2.00 mL的250 mg·L-1Cu2+溶液于10mL比色管中,用0.1mol·L-1氢氧化钠溶液和0.1mol·L-1盐酸溶液调节溶液酸度,考察了溶液酸度对Cu2+-西咪替丁配合物体系吸光度的影响。结果显示:当pH 小于7时,随着pH的增加,体系的吸光度随之增强;当pH 为7时,体系的吸光度最强且稳定;当pH 大于7时,体系的吸收光谱的峰形变宽,可能是由于部分Cu2+发生了羟基配位效应[21],从而影响与西咪替丁的主配位。因此,试验选用0.1 mol·L-1氢氧化钠溶液和0.1 mol·L-1盐酸溶液调节体系的酸度至pH 7。
试验进一步选用pH 7的乙酸-乙酸钠缓冲液来维持溶液酸度,并考察了pH 7的乙酸-乙酸钠缓冲液的用量对Cu2+-西咪替丁配合物体系吸光度的影响。结果显示:当pH 7的乙酸-乙酸钠缓冲液用量小于1.00 mL时,体系的吸光度随pH 7的乙酸-乙酸钠缓冲液用量的增大而增强;当pH 7的乙酸-乙酸钠缓冲液用量为1.00 mL 时,体系的吸光度最强;继续增大pH 7的乙酸-乙酸钠缓冲液的用量,体系的吸光度无明显变化。故试验选择pH 7的乙酸-乙酸钠缓冲液的用量为1.00 mL。
2.4 反应温度和反应时间的选择
固定其他条件不变,试验考察了反应温度对Cu2+-西咪替丁配合物体系吸光度的影响。结果显示:当反应温度小于60 ℃时,体系的吸光度基本保持不变;当反应温度大于60 ℃时,体系的吸光度随反应温度的增大而减小,且体系产生褐色沉淀,可能是由于在较高的反应温度下,Cu2+羟基配位效应增加,且生成的配合物发生了部分分解,从而导致吸光度降低[21]。因此,试验选择反应温度为室温。
试验考察了反应时间对西咪替丁与Cu2+络合反应的影响。结果显示,随着反应时间的延长,Cu2+-西咪替丁配合物体系的吸光度并无明显变化,说明反应时间对西咪替丁与Cu2+络合反应的影响不大。试验将反应时间控制在10 min。
2.5 试剂加入顺序的选择
保持其他条件不变,考察了试剂的加入顺序(顺序分别为:①西咪替丁标准溶液、乙酸-乙酸钠缓冲液、Cu2+溶液;②西咪替丁标准溶液、Cu2+溶液、乙酸-乙酸钠缓冲液;③乙酸-乙酸钠缓冲液、Cu2+溶液、西咪替丁标准溶液)对Cu2+-西咪替丁配合物体系吸光度的影响。结果显示:改变试剂的加入顺序,体系的吸光度变化不大。
2.6 干扰试验
西咪替丁片剂中含有淀粉、葡萄糖、羧甲基纤维素钠等辅料,试验考察了上述辅料及Al3+、Mn2+、Cr2+、Ni2+、Fe2+等金属离子对西咪替丁测定的影响。按上述优化后的条件配制溶液,以干扰物对西咪替丁测定结果的相对误差不超过±5%为标准。结果显示:2 950倍的葡萄糖,450倍的淀粉,120倍的Al3+,50 倍的羧甲基纤维素钠,25 倍的Mn2+,22倍的Cr2+,20 倍的Ni2+,2 倍的Fe2+不干扰测定;表明Fe2+、Ni2+、Cr2+、Mn2+对西咪替丁的干扰相对较大,但西咪替丁片样品和试验所用试剂均不含这些金属离子,故对试验不产生影响。
2.7 标准曲线和检出限
按照试验方法对质量浓度为50.47,100.9,151.4,201.9,256.0 mg·L-1的西咪替丁标准溶液系列进行测定。以西咪替丁的质量浓度为横坐标,Cu2+-西咪替丁配合物体系对应的吸光度为纵坐标绘制标准曲线。结果显示:西咪替丁的质量浓度在50.47~256.0 mg·L-1内,与其对应的Cu2+-西咪替丁配合物体系的吸光度呈线性关系,线性回归方程为A=1.926×10-3ρ+4.043×10-2,相关系数为0.998 4。
以3倍标准偏差(s)与斜率(k)的比值作为检出限(3s/k),计算该方法的检出限为3.00 mg·L-1。
2.8 精密度和回收试验
按照试验方法,对质量浓度为256.0 mg·L-1的西咪替丁标准储备溶液平行测定10次,计算测定值的相对标准偏差(RSD)。结果显示,测定值的RSD 为1.7%,表明方法的精密度良好。
按照试验方法对西咪替丁片样品溶液进行加标回收试验,以验证方法的可靠性,结果见表1。

表1 回收试验结果(n=5)Tab.1 Results of test for recovery(n=5)
由表1 可知,西咪替丁的回收率为95.3%~102%,测定值的RSD 为1.2%~2.9%。
2.9 稳定性和重复性试验
移取西咪替丁样品溶液4.00 mL,250 mg·L-1Cu2+溶液2.00 mL,pH 7的乙酸-乙酸钠缓冲液1.00 mL于10 mL 容量瓶中,加水稀释至刻度。以水为空白对照,分别于10,60,120,180,240,300,360 min时测定体系在325 nm 处的吸光度,计算测定值的RSD(n=7)为1.1%,表明Cu2+-西咪替丁配合物在360 min内基本稳定。
移取西咪替丁样品溶液4.00 mL,250 mg·L-1Cu2+溶液2.00 mL,pH 7的乙酸-乙酸钠缓冲液1.00 mL于10 mL 容量瓶中,加水稀释至刻度,平行配制6 份。以水为空白对照,在325 nm 处分别测定其吸光度,计算测定值的RSD(n=6)为1.6%,表明该方法重复性良好,符合分析测定的要求。
2.10 样品分析及方法比对
按照试验方法测定西咪替丁片剂1~3中西咪替丁的含量,结果见表2。
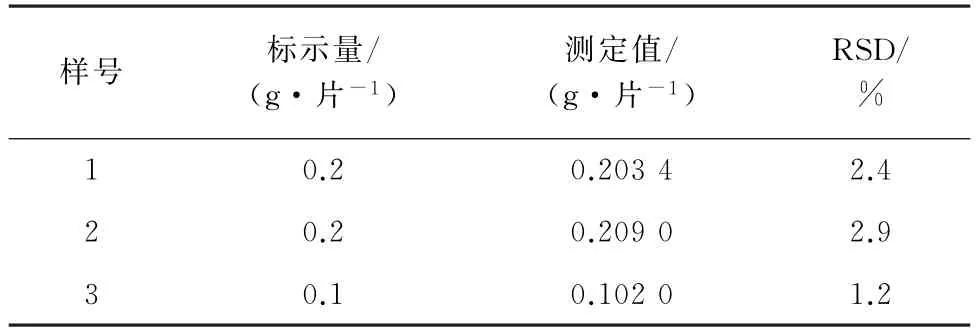
表2 样品分析结果(n=5)Tab.2 Analytical results of samples(n=5)
由表2可知,测定值与标示量基本一致,说明方法准确、可靠。
将本法与文献报道的方法进行对比,结果见表3。

表3 不同方法对比结果Tab.3 Comparison of results by different methods
由表3可知,本方法具有较宽的线性范围。
由于西咪替丁的紫外吸收波长位于218 nm 附近,存在溶剂末端吸收的干扰,会影响UV-Vis测定结果的准确度。本工作将西咪替丁和Cu2+络合,生成的Cu2+-西咪替丁配合物吸收波长红移至325 nm,通过测定Cu2+-西咪替丁配合物体系的吸光度,建立了UV-Vis间接测定西咪替丁含量的方法。该方法具有线性范围宽、操作快捷、环保低毒、结果准确的优点,并且将其成功运用到实际样品的测定,重现性好。
