基于土壤宏基因组分析的法医学个体来源地理位置推断
2021-08-20刘文丽程凤钱嘉林方晨刘旭范庆炜武会娟严江伟
刘文丽,程凤,钱嘉林,方晨,3,刘旭,3,范庆炜,武会娟,严江伟
1.北京市理化分析测试中心,北京 100089;2.山西医科大学法医学院,山西 太原 030001;3.北京市基因测序与功能分析工程技术研究中心,北京 100094;4.北京实验动物研究中心,北京100871
在法医学领域,土壤、水体等与案发现场相关的可疑物证应用于案件调查已有上千年的历史[1]。土壤检验可以揭示犯罪地点的信息,将嫌疑人或受害者与犯罪现场联系起来,为案件的侦破提供极为重要的线索和方向[2-3]。环境微生物学研究[4]表明,泥土、空气、水中分布着大量微生物,且其分布呈现多样性,不同地区、甚至同一地区不同地点的环境中微生物种类和构成均不相同。因此,可以根据个体可能携带的地理位置特异性微生物来推断其地域来源。
目前,对微生物群落的主要研究手段是宏基因组测序。该测序是一种不依赖培养,即可对环境样本中的所有微生物基因进行测序的方法,能鉴定复杂样本中微生物的构成[5],避免在培养过程中因污染或偏向性富集带来的偏差。细菌宏基因组的研究策略主要是全基因组鸟枪测序和16S rDNA基因测序。16S rDNA由高度保守的序列和相对高突变的序列相隔组成,保守区为细菌共有,可变区在不同细菌之间存在不同程度的差异,具有种属特异性,是微生物群落分析、进化及分类研究最常用的靶基因[6]。随着分子生物学和宏基因组技术的快速发展,人们对土壤微生物的认识不断加深,也推动了宏基因组学应用于法医学鉴定相关的土壤微生物多样性分析与群落构成鉴定[7]。
本研究采集了国内3 个省的33 例土壤样本,利用16S rDNA 高通量测序技术分析土壤的细菌菌群结构,比较不同地域来源的土壤菌落差异,为法医学个体来源地理位置推断提供科学依据。
1 材料与方法
1.1 实验样本
土壤样本采集自西藏自治区、青海省和黑龙江省,共33 例,按多点采样法取表层(0~10 cm)土壤,取200 g 混匀后作为该点的样本。其中西藏自治区(XZ)9 例,青海省(QH)12 例,黑龙江省(HLJ)12 例,样本位置信息如表1 所示。样本于-20 ℃中保存。

表1 样本信息Tab.1 Samples information
1.2 主要试剂和仪器
土壤基因组DNA 提取试剂盒[天根生化科技(北京)有限公司]、Qiagen 组织匀浆仪(德国Qiagen 公司)、Q5®High-Fidelity 2× Master Mix(美国NEB 公司)、KOD-Plus-Neo DNA 扩增酶(日本TOYOBO 公司)、Agencourt AMPure XP 产物纯化试剂盒(美国Beckman Coulter 公司)、QubitTMdsDNA HS Assay 试剂盒(美国Thermo Fisher Scientific 公司)、KAPA Li⁃brary Quantification 试剂盒(瑞士Roche 公司)、MiSeq Reagent 试剂盒v3(美国Illumina 公司)。
Miseq 测序仪(美国Illumina 公司)、7500型实时定量PCR 仪(美国Thermo Fisher Scientific 公司)、9700型PCR 仪(美国Applied Biosystems 公司)、Qubit®2.0荧光定量仪(美国Thermo Fisher Scientific 公司)等。
1.3 引物设计及合成
细菌16S rDNA V3-V4高变区PCR引物338F:5′-CCTACGGGNGGCWGCAG-3′;806R:5′-GGACTACH VGGGTWTCTAAT-3′;测序接头引物F:5′-AATGATA CGGCGACCACCGAGATCTACACT-3′;R:5′-CAAGC AGAAGACGGCATACGAGAT-3′,由生工生物工程(上海)股份有限公司合成。
1.4 DNA提取
土壤样本按照土壤基因组DNA 提取试剂盒的标准步骤进行提取,并对部分步骤进行改进:投入土壤样本为0.3 g;涡旋振荡采用Qiagen 组织匀浆仪,30 Hz 10 min;清洗DNA 后,加入30 μL 三 羟甲基氨基甲烷-乙二胺四乙酸[tris(hydroxymethyl)methyl amino⁃methane-ethylenediamine tetraacetic acid,TE]缓冲液洗脱DNA。
1.5 16S rDNA V3-V4高变区扩增
采用细菌16S rDNA V3-V4 高变区引物338F/806R,对细菌基因组DNA 及双蒸水作为阴性对照进行PCR 扩增。扩增体系:Q5®High-Fidelity 2× Master Mix 12.5 μL,10 μmol 上下游引物各1.25 μL,DNA 模板100 ng,无核酸酶水补至25 μL。PCR 反应条件:98℃30s;98℃10s,55℃30s,72℃30s,循环25次;72 ℃2 min。PCR 产物经1.2%琼脂糖凝胶电泳检测。
1.6 DNA测序文库构建及测序
V3-V4 高变区的PCR 产物利用KOD-Plus-Neo连接测序接头和标签序列。扩增体系:KOD-Plus-Neo 1 μL,10× PCR 缓冲液5 μL,2 mmol dNTP 5 μL,25 mmol MgSO43 μL,10 μmol 上下游引物各2 μL,PCR 产物4 μL,无核酸酶水补至50 μL。PCR 反应条件:94 ℃2 min;98 ℃10 s,55 ℃30 s,68 ℃30 s,循环9 次;68 ℃1 min。PCR 产物经1.2%琼脂糖凝胶电泳检测,评估构建的文库质量。用Agencourt AMPure XP产物纯化试剂盒进行文库纯化,使用KAPA Library Quantification 试剂盒对纯化后文库进行精确定量,最后使用MiSeq Reagent 试剂盒v3 和MiSeq 测序仪进行高通量测序,循环600 次,运行时间约65 h。
1.7 生物信息学分析
测序数据首先使用NGS QC Toolkit 软件[8]过滤掉低质量及低复杂度序列,再使用QIIME 软件[9]将相似度大于97%的序列归为1 个操作分类单元(opera⁃tional taxonomic unit,OTU)。基于Alpha 多样性指数,评估微生物群落的丰度和多样性,包括:利用丰富度指数(Chao1 指数和Observed_species 指数)评估样本中OTU 数目,数值越大,说明该样本物种数较多;利用测序深度指数(Goods_coverage 指数)计算样本的覆盖率,数值越大,则样本中序列被测出的概率越高;利用菌群多样性指数(Simpson 指数和Shannon 指数)评估每个样本的微生物多样性,数值越大,说明群落多样性越高。运用BLAST 算法比对Greengenes 数据库[10],完成OTU 的物种注释,选择检出量占85%以上的细菌进行各个水平下的分类分析。利用来自不同环境样品的OTU 代表序列构建OTU 系统发育树,再通过UniFrac 分析度量标准[11],利用各样本序列间的进化和丰度信息计算样本间加权和未加权的UniFrac距离,制作主坐标分析(principal co-ordinates analy⁃sis,PCoA)图以反映不同环境样本之间的微生物群落差异。
2 结果
2.1 16S rDNA V3-V4高变区扩增
PCR 产物的琼脂糖凝胶电泳检测结果见图1,33 个土壤样本V3-V4 高变区均得到较好的扩增,扩增片段单一,片段大小约465 bp,与目的片段大小一致;阴性对照扩增未出片段,扩增无污染,可进行后续实验。

图1 部分样本16S rDNA的V3-V4高变区扩增电泳结果Fig.1 Electrophoresis results of amplification of 16S rDNA V3-V4 hypervariable region of some samples
2.2 测序结果
本次测序构建3 个测序文库,共生成1.77 Gb 的测序数据,共获得2 720 149 条序列,序列平均长度约460 bp,每个样本序列数均>5 万条,可以进行后续的数据分析。
2.3 物种丰度
33 份样本的序列进行聚类,得到OTU 数目为114 848 个,基于OTU 结果计算丰富度指数,黑龙江省、青海省、西藏自治区土壤Chao1 指数平均值分别为797.45、745.11、535.98,Observed_species 指数平均值分别为91.83、88.25、84.22,土壤中细菌物种数黑龙江省最多,青海省次之,西藏自治区最少;测序深度指数(Goods_coverage 指数)各样本相差不大,测序深度基本覆盖到样本中的所有物种;多样性指数结果,黑龙江省、青海省、西藏自治区土壤Simpson 指数平均值分别为0.988、0.986、0.984,Shannon 指数平均值分别为6.46、6.36、6.25,土壤群落多样性从高至低依次为黑龙江省、青海省、西藏自治区(表2)。

表2 样品Alpha多样性指数Tab.2 Alpha diversity index of the samples
2.4 样本细菌群落结构
在门(phylum)水平上,黑龙江省土壤中占细菌检出量85%以上的门,含量从高到低依次为Proteo⁃bacteria、Actinobacteria、Acidobacteria、Verrucomicro⁃bia、Chloroflexi、Bacteroidetes、Planctomycetes;青海省土壤中占细菌检出量85%以上的门,含量从高到低依次为Planctomycetes、Acidobacteria、Gemmolatimonadetes、Proteobacteria、Bacteroidetes、Chloroflexi、Actinobacte⁃ria;西藏自治区土壤中占细菌检出量85%以上的门,含量从高到低依次为Proteobacteria、Bacteroidetes、Planctomycetes、Chloroflexi、Actinobacteria、Gemmolati⁃monadetes、Acidobacteria。在三省土壤样本中,均含有Proteobacteria、Actinobacteria、Acidobacteria、Chloro⁃flexi、Bacteroidetes、Planctomycetes 这6 个门,可见不同省虽然细菌种类有相同之处,但各门类的细菌分布却有差异(图2)。
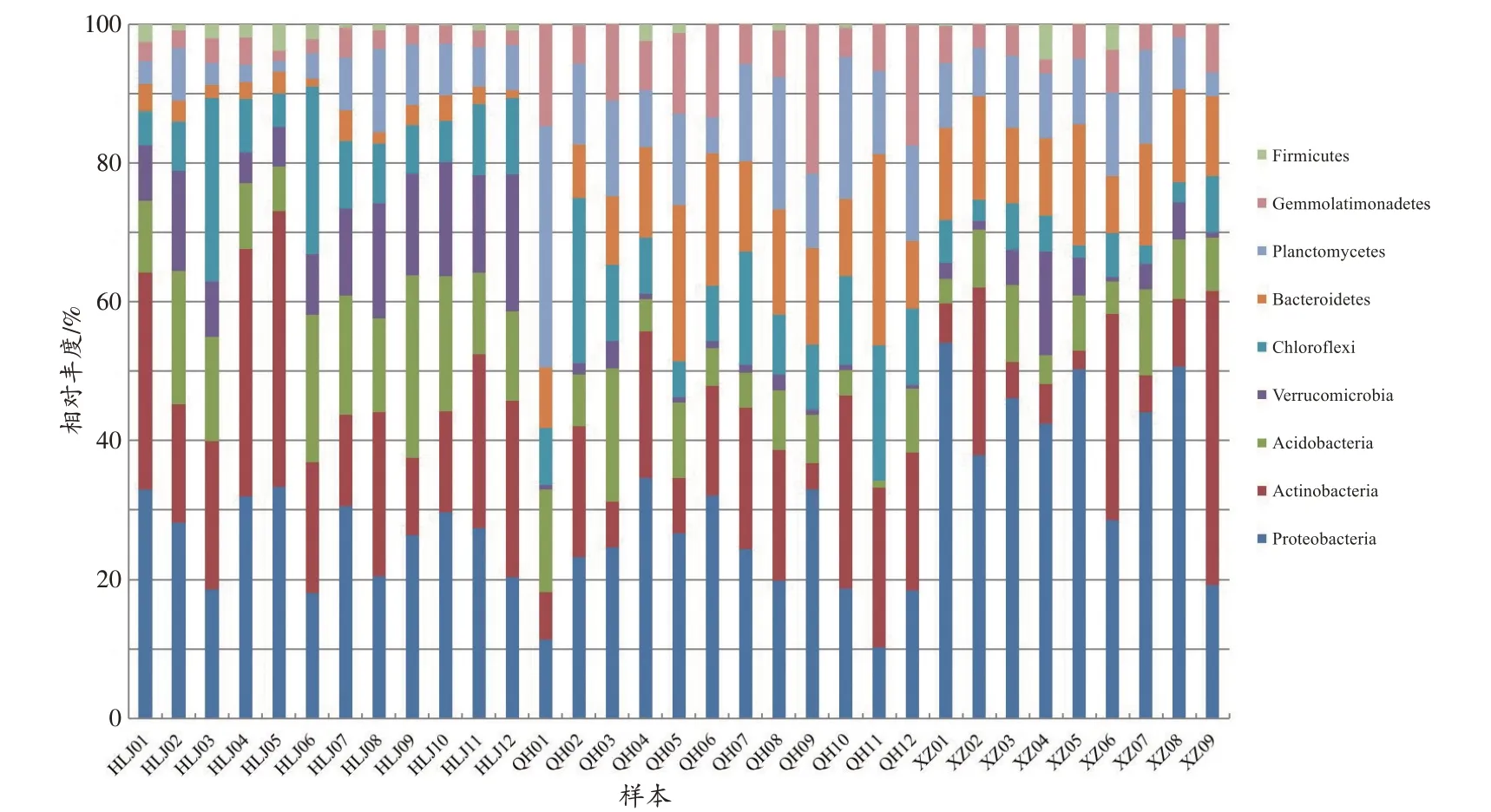
图2 33个样本在门水平的细菌组成Fig.2 Bacterial composition at phylum level in 33 samples
在属(genus)水平下,黑龙江省Verrucomicrobia门Chthoniobacteraceae 科的DA101属和Chloroflexi 门Thermogemmolatisporaceae 科某一未注释的属含量最高,为黑龙江省特有菌属。青海省Bacteroidetes 门Cytophagaceae 科某一未注释的属和Actinobacteria 门Nocardioidaceae 科某一未注释的属含量最高,为青海省特有菌属。西藏自治区Proteobacteria 门Comamona⁃daceae 科某一未注释的属和Verrucomicrobia 门Verru⁃comicrobiaceae 科Luteolibacter属含量最高,为西藏自治区特有菌属(图3)。

图3 3省样本在属水平的细菌组成Fig.3 Bacterial composition at genus level in 3 provinces’samples
2.5 PCoA
基于每个样本的所有OTU 相对丰度作PCoA 图(图4),按照加权UniFrac 度量分析结果为,主成分1的贡献率为32.36%,主成分2 的贡献率为16.35%,主成分3 的贡献率为7.65%,3 个主成分代表总变量的56.36%;按照未加权UniFrac 度量分析结果为,主成分1 的贡献率为19.76%,主成分2 的贡献率为9.11%,主成分3 的贡献率为5.94%,3 个主成分代表总变量的34.81%。以主成分1 和主成分3 为坐标轴构建的二维坐标系中,同一省土壤样本点的相互位置较近,可分别聚类到一个圈内,而不同省在PCoA 图上分布差异显著。

图4 加权和未加权PCoA结果Fig.4 Weighted and unweighted PCoA results
3 讨论
不同的地域有着不同的土壤环境,影响土壤细菌的数量以及种类分布特征。传统的法医学土壤检验主要是对颗粒大小、pH 值和含水量等理化特性进行分析[12]。但是根据这些特征值得出的结论可能因检测专家不同而不同,除非土壤中含有罕见的属性,否则对于地质上非常相似但来自不同环境下的样本,区分难度很大。因此,需要一种全面、客观的土壤鉴定和比较技术。随着二代测序技术的成熟和普及,对丰度极低的菌群也能进行有效分辨,可在短时间内获取微生物群落变化。使用细菌图谱来表征不同的土壤样本,为法医学土壤的个体化鉴定提供了可能[13]。
由于本研究为初步探索性研究,因此只选择了地理位置较远的西藏自治区和黑龙江省,以及与西藏自治区相邻的青海省,共3 个省的土壤样本。采用宏基因组研究方法,直接对采集的土壤样本进行细菌核酸提取和16S rDNA 的V3-V4 高变区扩增宏基因组学测序及生物信息学分析,在不依赖培养手段的情况下,在4 d 内即获取了土壤样本的菌群构成。测序共得到2 720 149 条测序序列,聚类得到的OTU 数目为114 848 个。基于OTU 结果计算丰富度指数及多样性指数,黑龙江省、青海省、西藏自治区土壤微生物Chao1 指数平均值分别为797.45、745.11、535.98,Observed_species 指数平均值分别为91.83、88.25、84.22;Simpson 指数平均值分别为0.988、0.986、0.984,Shannon指数平均值分别为6.46、6.36、6.25,结果表明,土壤中细菌物种数从多到少及群落多样性从高到低均依次为黑龙江省、青海省、西藏自治区,该结果也与王长庭等[14]的研究结果基本一致,细菌数量与植物群落地上生物量之间存在显著正相关。本研究的细菌群落分析结果表明,各省菌群既有种类相似性,又有差异性,黑龙江省优势菌属为Chthoniobacteraceae 科下的DA101属和Thermogemmatisporaceae 科下某一未注释的属;青海省优势菌属为Cytophagaceae 科下某一未注释的属和Nocardioidaceae 科下某一未注释的属;西藏自治区优势菌属为Comamonadaceae 科下某一未注释的属和Verrucomicrobiaceae 科下Luteolibacter属。未来在基于不同地区土壤大样本量微生物基因组数据的基础上,可考虑用不同地域土壤中的相对稳定和具有代表性的特定菌群作为法医学鉴定中指示地理地点相关信息的依据。PCoA结果表明,按照加权UniFrac 度量分析,3 个主成分代表总变量的56.36%,按照未加权UniFrac 度量分析,3 个主成分代表总变量的34.81%,不同省土壤细菌分布有显著差异,这是由于16S rDNA测序通量大、信息全、偏差小,因此不仅可区分与西藏自治区地理位置较远的黑龙江省,也可有效区分与西藏自治区地理位置较近的青海省。在未来,随着高通量低成本测序技术的广泛应用,全基因组测序能够提供除细菌外的全部微生物基因组信息,包括真菌、病毒等,从而更加深入全面地分析微生物构成和分布结构,不同地域土壤的区分度将进一步提升。
基于本研究,土壤样本可以根据细菌种类和分布特征而准确地进行区分或关联,从而给未知样本和嫌疑人的地理来源提供线索,也可将犯罪嫌疑人或物的样本与犯罪现场收集的样本进行比较,且不受空间和时间因素影响,因此细菌16S rDNA 测序在法医土壤分析和个体来源地理位置推断显示出巨大的潜力。但考虑到检测的准确性和可重复性,仍需进一步探讨样本的保存、DNA 提取、高通量测序等实验方法,同时加大测试的样本量,建立并完善相应的数据库、分析软件和判断标准,进一步证明该策略的可靠性。
