单倍型异基因造血干细胞移植治疗PRF1基因突变成人原发性噬血细胞性淋巴组织细胞增多症一例
2021-08-19孟广强张嘉王晶石王旖旎崔亭亭金志丽王昭
孟广强 张嘉 王晶石 王旖旎 崔亭亭 金志丽 王昭
患者,男,19岁,因“反复发热1年余”于2014年4月入院。患者于2013年4月无明显诱因出现发热,体温最高达40.2 ℃,伴畏寒,无寒战,伴乏力、全身肌肉关节酸痛。就诊于外院,查血常规示WBC计数3.07×109/L(3.5~9.5×109/L,括号内为正常参考值范围,以下相同),Hb 133 g/L(115~150 g/L),PLT计数21×109/L(125~350×109/L);白细胞分类见异型淋巴细胞;EB病毒(EBV)、巨细胞病毒(CMV)、呼吸道合胞病毒、腺病毒等抗体均为阴性;自身抗体相关检查阴性;腹部超声检查结果示肝脏及脾脏肿大、腹腔及腹膜后肿大淋巴结。考虑“传染性单核细胞增多症可能性大,淋巴瘤待除外”,予更昔洛韦抗病毒、甲泼尼龙治疗后,体温降至正常,血常规恢复正常后出院。2013年7月患者于劳累后再次出现上述症状,体温最高达39.0 ℃,再次就诊于外院查血常规示血小板减低。完善骨髓穿刺检查骨髓细胞学提示淋巴细胞占41%,其中异型淋巴细胞7%。免疫分型示:未见异常表型细胞。考虑“病毒感染”,予抗病毒治疗后体温恢复至正常水平。2013年10月前再次出现发热,体温最高达39.7 ℃,于外院复查骨髓细胞学示:可见大量疑似淋巴瘤细胞及较多吞噬细胞、吞噬血细胞现象。免疫分型示:未见明显异常表型细胞。IgH及TCR重排阴性。骨髓病理检查结果示:骨髓增生略低下,未见肿瘤细胞。正电子发射计算机断层显像(PET-CT)示:双侧颈部、双侧腋窝、纵隔、右肺门、肝门区、肠系膜区、腹膜后腹主动脉及下腔静脉周围、双侧腹股沟区多发高代谢大小不等淋巴结,骨髓呈弥漫性、局部结节样代谢增高,脾大伴代谢明显增高,考虑淋巴血液系统恶性病变可能。铁蛋白2 640 μg/L(11~306 μg/L)。sCD25>44 000 ng/L(<6 400 ng/L)。结合患者存在发热、肝脾肿大、中性粒细胞及血小板减少、铁蛋白升高、sCD25升高等特点,考虑诊断“噬血细胞性淋巴组织细胞增多症(HLH),淋巴瘤待除外”。2013年9月底予甲泼尼龙联合依托泊苷治疗,体温正常后糖皮质激素及依托泊苷逐渐减量至停用。2014年4月患者再次出现发热,为求进一步诊治来我院,复查血常规示:WBC计数2.50×109/L,Hb 148 g/L,PLT计数33×109/L。铁蛋白1 167.7 μg/L。EBV-DNA阴性。腹部超声检查示:脾脏肿大(厚7.7 cm)。自然杀伤细胞(NK细胞)活性11.37%(≥15.11%);sCD25>44 000 ng/L。复查骨髓细胞学示:增生明显活跃,淋巴细胞比例增高;骨髓有噬血现象。骨髓免疫分型示:未见明显异常细胞。骨髓病理检查结果示:未见肿瘤细胞。2014年4月予DEP方案(脂质体阿霉素+依托泊苷+甲泼尼龙)挽救治疗,过程顺利,患者出院后病情平稳,未再出现发热。送检HLH基因检测结果示:穿孔素1(PRF1)基因Exon3:c.1349C>T(p.T450M)纯合错义突变,且该突变与致病相关。追问其家族史,父母为近亲结婚。患者送检PRF1基因检测,结果示纯和错义突变c.1349C>T,导致所编码氨基酸发生突变(苏氨酸突变为甲硫氨酸)。患者父母行基因检测也存在PRF1基因杂合错义突变,且存在表达蛋白改变。而患者姐姐存在杂合同义突变,无表达蛋白改变(图1)。最终诊断:原发性HLH。配型检测提示其胞姐为HLA 5/10相合,后行单倍体异基因造血干细胞移植(allo-HSCT)。移植后检测未发现PRF1基因相关突变致病位点(图2)。此后患者定期门诊随诊。2020年9月门诊复诊,经评估HLH病情持续缓解状态。
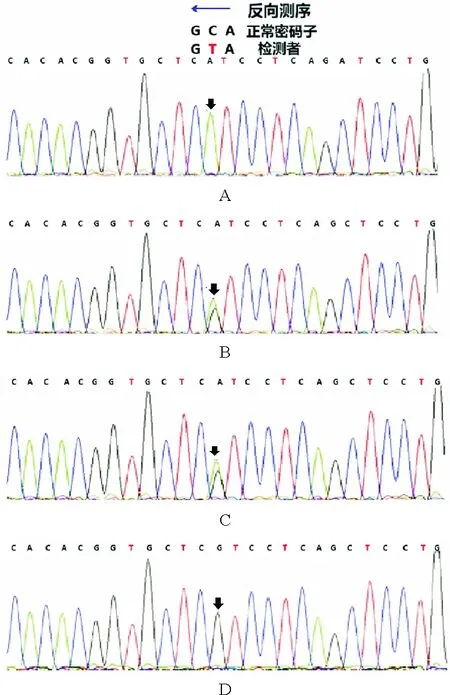
图1 基因测序图:患者PRF1基因Exon3:c.1349C>T(p.T450M)纯合错义突变,患者父母也存在PRF1基因杂合错义突变(A:患者;B:父亲;C:母亲:D:胞姐)
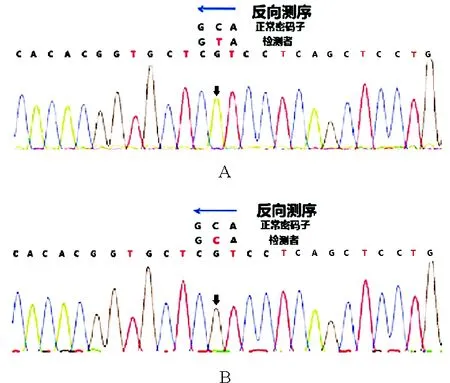
图2 患者移植前后基因改变:患者移植前PRF1基因Exon3:c.1349C>T(p.T450M)纯合错义突变;移植后检测未发现相关突变致病位点(A:移植前;B:移植后)
讨 论
HLH根据病因不同分为继发性HLH和原发性HLH。继发性HLH常见于感染、肿瘤和自身免疫性疾病。本例患者因反复发热,住院行相关检查后未发现明显感染和自身免疫性疾病方面的证据。虽曾怀疑淋巴瘤可能,但亦未发现病理学证据。而本病例因发现致病性PRF1基因突变最终诊断原发性HLH。原发性HLH是一种常染色体和(或)性染色体隐性遗传病,是由基因缺陷引起的NK细胞和细胞毒性T淋巴细胞(CTL)功能减低或缺如,从而导致过度免疫激活,产生大量炎症因子的过度炎症反应[1]。原发性HLH又分为家族性噬血细胞性淋巴组织细胞增多症(FHL)、免疫缺陷综合征及EB病毒驱动型HLH。本病例存在PRF1基因纯合错义突变c.1349C>T,为FHL-2。PRF1基因定位于染色体10q21-22,相关基因编码穿孔素蛋白,约占FHL的13%~50%[2-3]。当PRF1基因突变时,穿孔素的表达、活性及稳定性下降,此时受损的穿孔素无法顺利在靶细胞膜上形成管道,造成攻击细胞对靶细胞的杀灭作用受损,大量炎症因子累积失控,进而导致HLH。
基因筛查是诊断原发性HLH的金标准,目前的检测方法包括进行聚合酶链反应(PCR)产物直接测序及高通量DNA测序技术。对考虑原发的HLH患者应该尽早完善基因筛查,以明确原发HLH诊断和指导治疗。除了基因筛查外,以流式细胞术为基础的功能学检测近来作为原发HLH的快速筛选方法。穿孔素、颗粒酶B、SAP、XIAP和Munc13-4表达均可以通过细胞内着色来检测,相关结果可提示可能存在FHL-2、XLP-1、XLP-2及FHL-3型HLH。对于怀疑原发性HLH的患者,可以给予先行流式细胞术进行上述检测以快速筛选原发HLH。但是该方法并不能取代基因检测,基因筛查仍是金标准。因此,对于反复复发、未发现明确病因者,应积极行相关基因的检测以明确有无原发性HLH的可能。本病例应用糖皮质激素和依托泊苷疾病缓解后反复复发,而且需要特别指出的是本病例父母为近亲婚配。以上均提示本病例非常必要行原发性HLH的基因检测。最终基因检测结果示PRF1基因Exon3:c.1349C>T(p.T450M)纯合错义突变,且该突变目前已被证实与致病相关,而本病例父母也存在PRF1基因杂合错义突变。最终行姐供弟单倍体allo-HSCT,移植后基因检测未再发现相关致病基因突变位点。
allo-HSCT是目前认为唯一可以治愈原发性HLH的治疗方法。allo-HSCT自1986年首次报道用于治疗HLH至今已有30余年[4]。allo-HSCT在儿童HLH治疗中的重要地位已得到充分证实。如在HLH-04研究中,接受allo-HSCT的133例原发HLH患儿的移植后5年总生存(OS)率为70%;而未接受移植的35例儿童中仅有2例生存[5]。目前allo-HSCT治疗成人原发性HLH的报道较少,且多为个案报道和小样本研究,但已被证实是成人原发性HLH的有效治疗方法[1,6]。对于原发性HLH的亲缘供者选择需慎重,因为亲属可能携带相同的基因突变。虽然目前尚无证据证实亲缘同胞如父母携带的等位基因突变会增加患者移植后再次发生HLH的风险[7],但为避免这种情况的发生,应尽量选择无HLH相关等位基因突变供者。最终我们选择无该基因突变的胞姐作为供者。本例患者自allo-HSCT至今已有6年余,一般状况良好。结合该患者治疗过程,我们认为单倍型allo-HSCT是成人原发性HLH治愈的重要方法。
