青少年起病的成人型糖尿病合并背侧胰腺发育不全一例
2021-08-19束燕雯顾天伟陆婧沈山梅毕艳朱大龙
束燕雯 顾天伟 陆婧 沈山梅 毕艳 朱大龙
患者,女,31岁,因“发现血糖升高11年”于2018年8月17日入院。患者11年前因下肢疖痈于外院就诊输液治疗时出现腹痛、呕吐、昏迷,末梢血糖测不出,糖化血红蛋白(HbA1c)15.2%,血气分析结果提示酸中毒,诊断为“酮症酸中毒,1型糖尿病”,予四针胰岛素方案(门冬胰岛素注射液早/中/晚餐前10 U/8 U/8 U联合甘精胰岛素睡前14 U)降糖治疗,用药期间出现低血糖(具体不详)。后改用门冬胰岛素30早餐前22 U皮下注射、阿卡波糖50 mg早餐时服用,仍频发低血糖,多发生于清晨。此次为进一步调整血糖水平至我院就诊。既往史:有糖尿病视网膜病变1期、甲状腺结节、慢性非萎缩性胃炎,无胰腺炎及胰腺手术病史。患者系第一胎、第一产,足月顺产,出生时体重3.8 kg,出生时无低血糖史,母亲妊娠期未发现糖尿病,母乳喂养,生长发育与同龄人相当。婚育史:未婚未育。家族史:患者母亲、舅舅、外婆及舅祖父均有糖尿病;患者母亲47岁时诊断糖尿病,不规律口服格列齐特缓释片降糖,平素空腹血糖控制在5 mmol/L左右,餐后2小时血糖未规律监测;患者舅舅60岁为诊断糖尿病,具体治疗及血糖控制情况不详;患者外婆70岁为诊断糖尿病,同年因心脏疾病去世(具体不详);患者舅祖父糖尿病起病年龄不详,因心脏疾病去世,患者家系系谱图见图1。入院后体格检查:T 36.3 ℃,P 67次/分,R 14次/分,Bp 121/85 mmHg,身高160 cm,体重60 kg,BMI 23.4 kg/m2,腰围70 cm,臀围90 cm,腰臀比0.78,无腹型肥胖,无库欣貌,无黑棘皮症,心、肺、腹部检查未见明显异常,双下肢无水肿。辅助检查结果:空腹血糖(FPG)4.6 mmol/L(3.9~6.1 mmol/L,括号内为正常参考值范围,以下相同),标准餐后2小时血糖(2h PG)7.9 mmol/L(<11.1 mmol/L);空腹C肽144.1 pmol/L(370.0~1 470.0 pmol/L),餐后2小时C肽502.3 pmol/L;HbA1c 6.5%(4.2%~6.0%);胰岛自身抗体检查结果均为阴性。甘油三酯0.32 mmol/L(0.56~1.70 mmol/L),低密度脂蛋白胆固醇2.41 mmol/L(1.89~3.10 mmol/L),肝肾功能检查结果未见明显异常。眼底照相检查结果示:糖尿病视网膜病变Ⅱ期;尿微量白蛋白/肌酐7 mg/g(0~30 mg/g)。踝肱指数(ABI)、颈动脉超声、心脏超声、肌电图检查结果均未见明显异常。泌汗功能测试检查结果提示怀疑有考虑不对称的外周自主神经病变。72小时动态血糖监测(门冬胰岛素注射液早/中/晚餐前10 U/8 U/8 U联合甘精胰岛素睡前14 U)结果显示:共测定血糖值852个,血糖波动在2.5~14.2 mmol/L,平均值6.0 mmol/L,标准差2.7 mmol/L,变异系数44.2%,平均葡萄糖波动幅度(MAGE)5.8 mmol/L;午夜至凌晨(23∶00~6∶00)反复出现低血糖,血糖水平≤3.9 mmol/L及≤2.8 mmol/L的时间(百分比)分别为15小时5分钟(21%),0小时55分钟(1%),见图2A。根据MODY概率计算器(MPC)[1]测算,患者的MODY阳性预测值为49.4%,建议进一步行基因检测筛查单基因糖尿病。二代测序检查结果提示:位于第20号染色体的肝细胞核因子4α(HNF4α)基因第6外显子编码区(NM_000457)产生突变c.671C>A(编码区第671号核苷酸由胞嘧啶变异为腺嘌呤),导致氨基酸改变p.A224D(第224号氨基酸由丙氨酸变异为天冬氨酸),为错义突变。该突变在正常人群数据库中的频率为0,为低频变异;生物信息学蛋白功能预测软件SIFT、PolyPhen2、REVEL 分别预测均为有害;HGMD数据库无该位点的相关性报道;患者母亲存在相同突变(图3)。根据基因检测结果,修正诊断为HNF4α-MODY。2018年11月1日患者再次入院拟调整治疗方案,完善检查胰腺CT平扫+增强检查结果显示:胰体尾部显示不清;影像学诊断为背侧胰腺发育不全,见图4。再次修正诊断为“HNF4α-MODY合并背侧胰腺发育不全”。调整降糖方案:停用胰岛素,改为格列齐特缓释片从15 mg每日1次逐渐增加剂量至45 mg每日1次。复查动态血糖监测 (格列齐特缓释片每日1次):共测定血糖值579个,血糖波动在3.7~7.3 mmol/L,平均值5.2 mmol/L,标准差0.7 mmol/L,变异系数14.1%,MAGE为1.8 mmol/L,血糖水平≤3.9 mmol/L及≤2.8 mmol/L的时间(百分比)分别为2小时0分钟(4%),0小时0分钟(0%),见图2B。患者出院后监测餐后血糖偏高,因此调整治疗方案为格列齐特缓释片60 mg每日1次+阿卡波糖片50 mg每日3次。此后患者分别于2019年1月、2019年9月来我院随访,查HbA1c 6.4%,自诉血糖控制可,未发生低血糖,胰岛功能随访结果见表1。
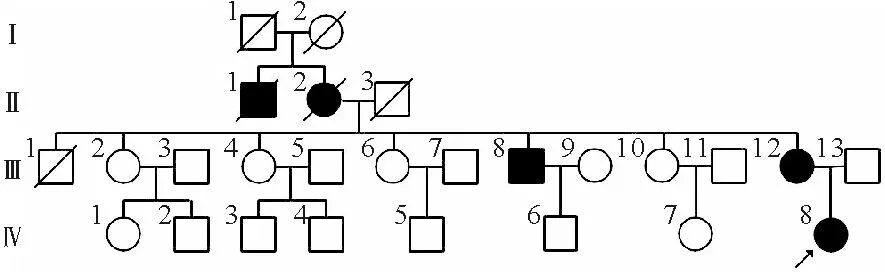
图1 患者家系系谱图(箭头表示先证者;圆圈表示女性,方框表示男性,空心表示血糖正常,实心表示临床诊断糖尿病,斜杠为去世个体,先证者为Ⅳ-8,其中Ⅲ-12与先证者存在相同突变,余家系成员未进行基因检测)
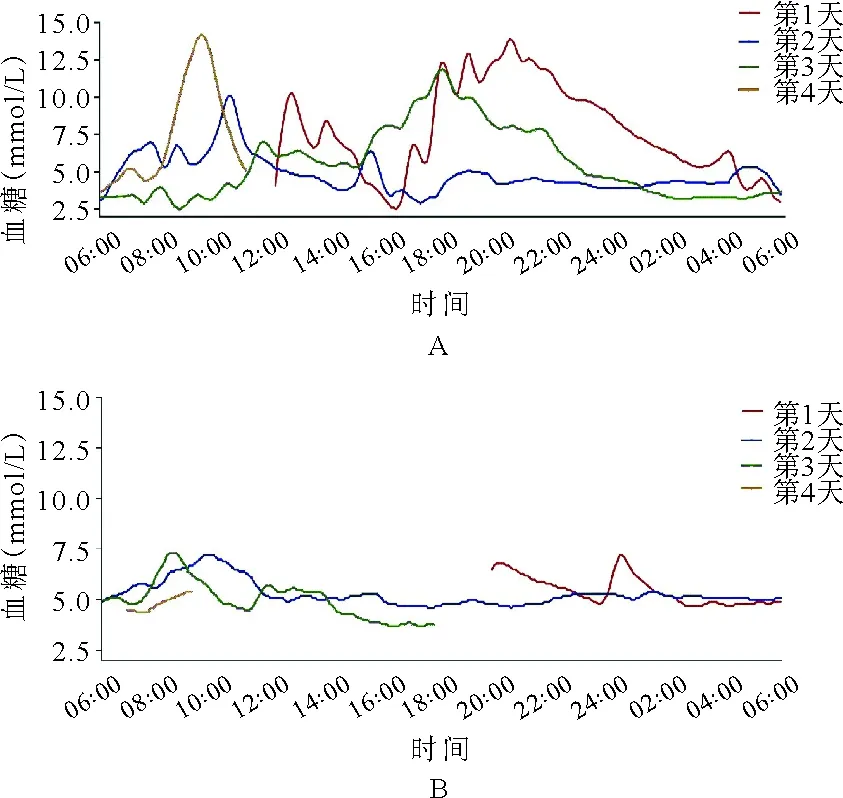
图2 患者动态血糖监测结果(A:入院时;B:调整治疗方案后)

图3 二代测序分析结果(A:患者HNF4A 基因第6外显子c.671C>A杂合突变;B:患者母亲HNF4A基因第6外显子c.671C>A杂合突变)
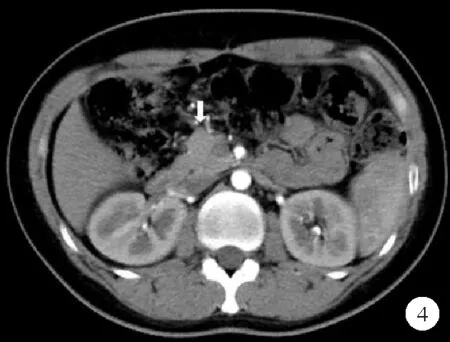
图4 患者胰腺CT增强检查结果(胰腺体尾部缺如,胰腺呈球状,箭头所指为胰头)
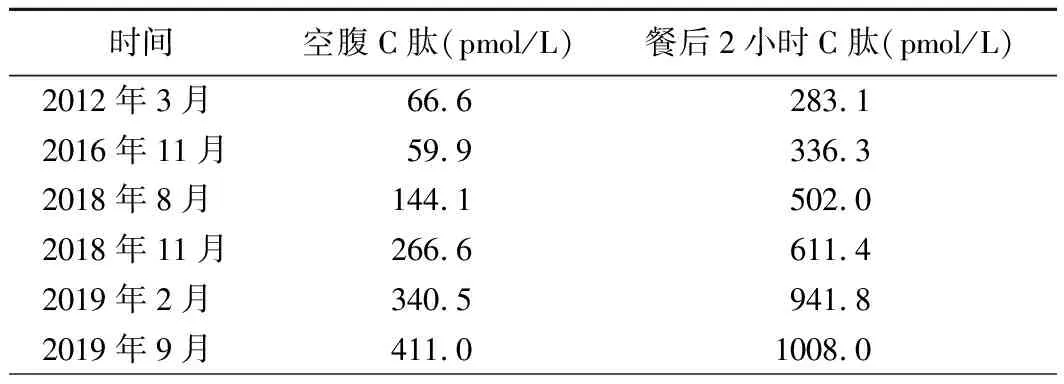
表1 患者胰岛功能变化情况
讨 论
本病例首次报道了HNF4α-MODY合并背侧胰腺发育不全。MODY是单基因糖尿病中的一类,其致病基因编码的蛋白中既有与胰岛β细胞分化、发育和分泌胰岛素功能密切相关的转录因子,又有参与糖、脂代谢的关键酶和调控胰岛β细胞分泌胰岛素的K+-ATP通道的两种亚单位组成蛋白[2]。临床诊断标准为:(1)有连续三代及以上的家族史,呈常染色体显性遗传;(2)家族中至少1人发病年龄<25岁;(3)至少5年不依赖胰岛素治疗,无酮症倾向[3]。但以此标准可能会漏诊大量患者,为提高诊断效率,Shields等[1]提出了基于基本临床信息的诊断模型并提供了在线分析计算器,但最终确诊及分型诊断仍有赖于基因检测结果。HNF4α-MODY在目前已知的14个MODY亚型中较为常见,仅次于GCK-MODY及HNF1α-MODY,1996年至今共发现超过94个HNF4α-MODY家系[4],但在亚洲人群中HNF4α-MODY分布较少,在中国人群中更为罕见。HNF4α-MODY由HNF4α突变所致,HNF4α不仅通过影响其他转录因子表达调节胰岛素基因的转录,还影响参与葡萄糖转运及代谢相关酶的转录[5],其突变致HNF4α-MODY的潜在遗传学基础主要为单倍体不足,单个等位基因的功能缺乏可导致糖尿病表型[6]。除MODY的共同特征外,HNF4α-MODY患者还具有以下特点:(1)由宫内胰岛素分泌增加导致的巨大儿及新生儿低血糖[7];(2)因影响脂代谢导致的甘油三酯水平较低[3];(3)大血管及微血管并发症和1型糖尿病及2型糖尿病相近;(4)对磺脲类药物敏感,但随着病程进展,胰岛功能逐步衰退,最终仍需加用胰岛素治疗。本例先证者有明确糖尿病家族史,17岁时因糖尿病酮症酸中毒起病,初诊为“1型糖尿病”,应用四针胰岛素及预混胰岛素治疗过程中血糖波动均较大,且多次发生低血糖,2018年入我院检查胰岛功能未见明显衰竭,胰岛自身抗体阴性。为明确分型诊断,进一步行基因检测提示HNF4α-MODY。HNF4α-MODY患者起病时多呈现胰岛功能轻度减退,因此发生酮症或酮症酸中毒的几率较低,但既往亦有文献报道MODY患者可发生酮症酸中毒[8],尤其在合并应激状态时,如本例患者合并下肢皮肤感染。因此,酮症起病并不是MODY的排除条件,对于临床表现不典型的“1型糖尿病”患者,需考虑MODY可能性。
背侧胰腺发育不全是一种以胰腺颈体尾部未发育或发育不全的先天性胰腺发育畸形,多数患者无临床症状,部分患者表现为腹部疼痛[9],也可出现葡萄糖代谢异常,甚至合并糖尿病酮症酸中毒[10]。其诊断主要依靠影像学检查,特征为胰腺仅有头部残留(可增大、正常或缩小),胰腺体尾部部分缺失[11],中国人群中亦罕见报道。调控胰腺发育的基因按胰腺发育顺序可分为3大类:hcf3、pdx-1和hlxb9等决定胰腺发生的基因;ngn3、isl1和p48等决定胰腺细胞定向分化的基因;Nkx2.2、Nkx6.1、Pdx-1、isl1、Bh9、Pax4、Pax6、Brn4 和 NeuroD1等决定内分泌细胞世系分化的基因[12]。有研究结果显示,胰腺发育不全与 GATA6、PDX1、PTF1A、GATA4、RXF6和HNF1B 等基因突变相关[13]。我们的候选基因中已包括胰腺发育相关的GATA6、PDX1、PTF1A基因,并未发现突变。该患者是否存在其他未筛查基因的突变尚不得知,仍需行全外显子测序甚至全基因组测序才能进一步明确,但患者拒绝行进一步基因检查。此外,由于患者母亲未提供胰腺CT检查结果,因此并无信息表明患者母亲是否同样合并胰腺发育不全。患者存在HNF4α突变,HNF4α在肝、肾、肠和胰岛中均有表达,不仅调节与葡萄糖、脂肪酸及胆固醇代谢相关的基因,在个体发育中也具有重要的调节作用,小鼠敲除HNF4α后甚至可致胚胎死亡[14]。目前尚未见文献报道HNF4α基因突变与背侧胰腺发育不全有关,该患者的胰腺发育不全是否与HNF4α对调控胰腺发育的基因的调节作用有关,仍需待进一步证实。
本例患者病程较长且合并背侧胰腺发育不全,入院时实验室检查结果提示胰岛功能欠佳,但尚未完全衰竭。有研究对HNF4α-MODY家系(R-W家系)进行36年随访,最终仅30%的患者需要进行胰岛素治疗,部分HNF4α突变患者在磺脲类药物治疗33年后,葡萄糖刺激下胰岛素分泌水平仍有显著升高,这些低剂量长期磺脲类药物治疗的有效性提示,HNF4α-MODY和HNF1α-MODY患者大多对磺脲类药物敏感[15]。但对于未被报道过的突变,在调整治疗方案时,仍需严密监测血糖以观察疗效,如p.R114W突变患者对磺脲类药物敏感性较差[16]。英国一项前瞻性研究指出,对于病程较长(>11年)的HNF4α-MODY患者,可在现有治疗基础上加用磺脲类药物,体现了联合治疗的重要性[16]。对于HNF4α-MODY患者,可考虑联用二肽基肽酶4(DPP-4)抑制剂及胰高血糖素样肽1(GLP-1)受体激动剂[18]。其中DPP-4抑制剂阿格列汀在HNF4α-MODY患者的有效性和安全性已有相关文献报道[19],GLP-1受体激动剂对部分HNF4α-MODY患者的益处亦有报道[20],而钠-葡萄糖协同转运蛋白2(SGLT-2)抑制剂由于其作用不依赖于胰岛素,可在联合治疗中发挥作用[21],但其安全性及有效性尚需足够的数据支持。一般情况下,初发MODY患者呈现胰岛功能轻度减退,而本例患者胰岛功能明显低下,考虑与其合并胰腺的先天畸形有关。目前该患者接受磺脲类药物联合阿卡波糖治疗后HbA1c达标,且较前更为平稳,低血糖事件较前减少,经过10个月的随访胰岛功能略有改善,但仍需继续长期随访评估疗效。
综上,本文通过对1例HNF4α-MODY合并背侧胰腺发育不全患者的临床特征及治疗转归进行分析,为其临床诊治提供一定参考。MODY易被延迟诊断及误诊、漏诊,尽早识别MODY并给予个体化治疗,对于改善患者血糖、提高生活质量尤为重要。
