Zr-MOF空心纳米球固载离子液体对CO2环加成反应的催化性能
2021-08-16李美燕陈紫娟汪淑华
李美燕,陈紫娟,汪淑华,陈 超
(南昌大学化学学院,江西环境与能源催化重点实验室,南昌 330031)
二氧化碳(CO2)既是一种主要的温室气体,也是一种重要的碳资源,以CO2为原料合成高附加值的化学品已吸引了全世界研究者的关注[1~3].其中CO2和环氧化物发生环加成反应生成环状碳酸酯的反应绿色环保,且产物被广泛应用于生物医药和化工等领域,被认为是最有效的CO2资源化利用的途径之一[4~6].目前已开发出多种催化剂催化该反应,其中,由有机阳离子和有机或无机阴离子组成的在室温下呈液态的离子液体[7,8],如咪唑盐[9]、季铵盐[10]、季鏻盐[11],具有催化性能高、可设计性强且绿色环保等优点,被作为一种新型绿色催化剂广泛应用于催化CO2环加成反应.Zhang等[12]和Luo等[13]设计并制备了羟基功能化季铵盐等一系列功能化离子液体,并证明羟基和羧基等氢键给体能与环氧化合物形成氢键,与卤素阴离子协同作用,促进环氧化物开环活化,降低反应的活化能,从而使反应更容易进行.但是离子液体作为一种黏度高的均相催化剂,很难从催化体系中分离,限制了其实际应用,将离子液体固载到金属-有机框架(MOFs)等多孔材料中是解决这个问题的有效方法之一.
MOFs是由金属离子和有机配体通过配位自组装形成的周期性网状多孔材料,具有比表面积大、金属位点开放和结构多样性等特性[14,15],通过改变有机连接剂和金属配合物,可很容易地调整MOFs的孔径、形状和气体亲和力,从而调节配体和金属中心的各种功能[16~18].且含有不饱和金属配位点的MOFs,作为Lewis酸性多相催化剂能有效催化转化CO2[19,20].然而,大多数MOFs材料作为催化剂时需要在高温、高压或者助催化剂的存在下才能有效催化转化CO2,这将大大增加反应成本.使用MOFs固载离子液体,有助于提高MOFs的催化活性,同时也解决了离子液体作为均相催化剂难回收等问题.Aguila等[21]利用自由基聚合在MIL-101的孔洞内产生线性离子液体聚合物(IP),并将MIL-101-IP应用于CO2环加成反应中,其在常温常压下表现出非常高的活性,但由于传质效率低下导致反应时间长达48 h,时间成本和能耗成本较大,限制了其工业化应用.研究证明,中空纳米结构材料具有密度小、传质效率高等特点[22],本课题组[23]报道的空心双壳层MOFs材料(Void@HKUST-1@Pd@ZIF-8)进一步证明了这一点,与实心MOFs相比,中空结构的Void@HKUST-1@Pd@ZIF-8应用于加氢反应时拥有更高的传质效率和反应活性.基于此,本文合成了空心纳米球和实心块2种结构的季铵盐功能化UiO-66-NH2,并将其应用于催化CO2和氧化苯乙烯的环加成反应.通过卤素阴离子、—OH和中空结构的协同作用,空心纳米球Void@UiO-66-Ⅰ催化剂表现出比实心块UiO-66-Ⅰ催化剂更好的催化活性.
1 实验部分
1.1 试剂与仪器
五水合硝酸锆(纯度98%,阿达玛斯试剂有限公司);氯化锆(分析纯,阿拉丁试剂有限公司);2-氨基对苯二甲酸(NN2-BDC,分析纯,毕得医药股份有限公司);聚乙烯吡咯烷酮k30(PVP,优级纯,国药集团化学试剂有限公司);碘乙醇(分析纯,阿达玛斯试剂有限公司);N,N-二甲基甲酰胺(DMF)、过硫酸钾、浓硫酸、乙酸、苯乙烯、无水乙醇和无水甲醇均为分析纯,购于国药集团化学试剂有限公司;氧化苯乙烯(SO,纯度98%,安耐吉化学有限公司);去离子水为自制.
SmartLab9 kW型X射线衍射仪(XRD,日本理学公司);Autosorb-iQ型物理吸附仪(美国康塔仪器公司);ESCALAB 250Xi型X射线光电子能谱仪(XRD,美国赛默飞世尔科技公司);AVANCEⅢHD 400 MHz型核磁共振波谱仪(NMR,德国布鲁克公司);Various EL-3型有机元素分析仪(德国Elementar公司);SUB100型环境扫描电子显微镜(SEM,日本日立集团);JEM-2100型透射电子显微镜(TEM,日本电子株式会社);Agilent 5100型电感耦合等离子体发射光谱仪(ICP,美国安捷伦科技有限公司);DSCTGA3+型热重分析仪(瑞士梅特勒-托利多仪器公司);Nicolet iS10型傅里叶变换红外光谱仪(FTIR,美国赛默飞世尔科技公司);7820A型气相色谱仪(GC,HP-5柱:30μm×20 mm×0.25 mm)和GC7890AMS5975型气质联用(美国安捷伦科技有限公司).
1.2 实验过程
1.2.1 UiO-66-Ⅰ的合成 UiO-66-NH2的合成:将1.8 g氯化锆分散于20 mL DMF中,在室温下搅拌均匀.在另一个烧杯中,加入1.27 g 2-氨基对苯二甲酸、0.65 mL氨水(2 mol/L)和20 mL DMF,并搅拌均匀.将制备好的2-氨基对苯二甲酸溶液缓慢地滴加入氯化锆溶液中,滴加结束后,加入60 mL DMF并继续搅拌均匀.随后升温至110℃,反应24 h.反应结束后用DMF洗涤,并用无水甲醇交换3 d,真空干燥得到UiO-66-NH2.
UiO-66-Ⅰ的合成:通过后合成修饰方法(PSM)合成UiO-66-1.将0.5 g UiO-66-NH2真空活化后,分散于碘乙醇中,边搅拌边向体系内通入氮气15 min,排出空气.将温度升高至60℃,在氮气保护下反应4 d.由于烷基化是逐步进行的,所以要使用过量的碘乙醇与UiO-66-NH2重复反应几次,即第一次反应过后,离心收集UiO-66-Ⅰ-1,用无水甲醇洗涤后真空活化,再重复上述操作,得到UiO-66-Ⅰ-2,UiO-66-Ⅰ-3和UiO-66-Ⅰ-4.反应结束后,用无水甲醇洗涤并交换3 d,之后在真空干燥箱中干燥得到UiO-66-Ⅰ.合成过程如Scheme 1所示.
1.2.2 Void@UiO-66-Ⅰ的合成 磺化聚苯乙烯小球(SPS)的合成:聚苯乙烯(PS)小球的制备以及磺化参考文献[24]方法进行.
SPS@UiO-66-NH2的合成:根据文献[25]方法合成UiO-66.在100 mL烧瓶中,加入0.5 g上述合成的SPS小球、0.5 g PVP、2.23 g五水合硝酸锆(5.2 mmol)、0.9 g 2-氨基对苯二甲酸(5 mmol)和10 mL乙酸,以水为溶剂,在100℃下反应24 h.反应结束后,通过离心收集产物,并用去离子水和无水甲醇洗涤3遍,在真空干燥箱中干燥后得到SPS@UiO-66-NH2.
SPS@UiO-66-Ⅰ的合成:SPS@UiO-66-Ⅰ的合成步骤与UiO-66-Ⅰ一致,在合成过程中将UiO-66-NH2换成SPS@UiO-66-NH2,最终得到产物SPS@UiO-66-Ⅰ.
Void@UiO-66-Ⅰ的合成:将SPS@UiO-66-Ⅰ分散于DMF中,浸泡2 d,每天更换3次DMF,然后用无水甲醇交换3 d,每天更换新鲜的无水甲醇,交换完成后真空干燥得到Void@UiO-66-Ⅰ.合成过程如Scheme 1所示.

Scheme 1 Preparation process for UiO-66-I and Void@UiO-66-I
1.3 催化剂的活性测试
催化剂每次使用前均经过真空活化.CO2和环氧化合物反应的操作步骤为:将25 mmol氧化苯乙烯(SO)和0.5%(摩尔分数)活化后的催化剂加入到聚四氟乙烯内衬的高压反应釜中,加入磁力搅拌子,将高压反应釜密封,用CO2(纯度为99.99%)除净釜内空气后,充入一定压强的CO2,升温,磁力搅拌反应一定时间后,将高压反应釜转移至冰水混合物中,快速终止反应,将釜内多余的CO2排出,通过离心分离得到催化剂和反应液.催化剂用无水甲醇洗涤后,再交换3 d,每天更换甲醇以除去催化剂中残余的反应物或产物分子.反应液用丙酮稀释后,通过GC进行分析,计算转化率和选择性.
2 结果与讨论
2.1 催化剂的表征
对被修饰后的MOFs材料进行了XRD测试,结果如图1所示.由于季铵盐部分是该催化剂的主要活性位点,所以在框架内尽量多的引入季铵盐有助于提高其催化活性,但这一点必须在MOFs结构保持完整的基础上进行.由图1(A)可见,UiO-66-NH2与碘乙醇反应4次后,在2θ=4.5°左右出现了一个新的特征衍射峰.据文献[25]报道,当MOFs内部产生了有序的晶体缺陷时,会在其XRD谱线中出现新峰.因此样品的XRD谱图出现新峰可能是因为引入了过多的季铵盐导致部分MOFs孔道破损,产生了晶体缺陷,这不利于材料的稳定性,因此催化剂的最佳反应条件为碘乙醇与UiO-66-NH2反应3次,并将UiO-66-Ⅰ-3简记作UiO-66-Ⅰ.通过图1(B)可见,对SPS@UiO-66-NH2进行修饰后,其XRD特征峰强度略有减弱,但整体峰型没有明显变化;将模板去除后,XRD特征峰强度整体增强,表明形成了中空结构且MOFs骨架没有坍塌.
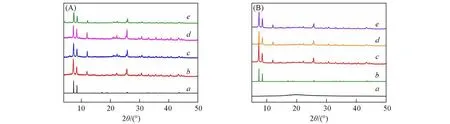
Fig.1 XRD patterns of UiO-66-I-n(A)and each step product of Void@UiO-66-I catalyst(B)(A)a.Simulated UiO-66;b.UiO-66-Ⅰ-1;c.UiO-66-Ⅰ-2;d.UiO-66-Ⅰ-3;e.UiO-66-Ⅰ-4.(B)a.SPS;b.simulated UiO-66;c.SPS@UiO-66-NH2;d.SPS@UiO-66-Ⅰ;e.Void@UiO-66-Ⅰ.
为确认MOFs的后合成修饰是否成功及其修饰程度,对催化剂进行了FTIR,XPS,1H NMR以及元素分析等表征.首先对UiO-66-NH2,UiO-66-Ⅰ-n(n=1,2,3),Void@UiO-66-NH2和Void@UiO-66-Ⅰ进行了FTIR测试(见本文支持信息图S1).1625 cm-1处的吸收峰可归属于伯胺的N—H弯曲振动混合,随着使用碘乙醇对UiO-66-NH2修饰次数的增加,红外吸收峰逐渐减弱[26];与苯环连接的CAr—N键的吸收峰发生了偏移,由原来的1255 cm-1变为1286 cm-1[27],且在1080 cm-1处出现了新的吸收峰,这与文献[28]报道的C—O特征峰一致,上述结果表明,MOFs的后合成修饰成功,且随着反应次数的增加,参加反应的UiO-66-NH2的氨基数量在增加.但还不能确定是否有季铵盐在UiO-66内生成,进一步对材料进行了XPS测试,结果示于图2.可见,UiO-66-NH2和Void@UiO-66-NH2的N1s谱图均只有一个特征峰,即在399.65 eV处呈现的胺氮N1s特征峰[图2(A)和(A′)],I3d谱图中未见任何特征峰,表明修饰前样品中不存在碘元素[图2(B)和(B′)].季铵化反应后,UiO-66-Ⅰ和Void@UiO-66-Ⅰ的N1s谱图中除了原来的胺氮N1s特征峰,还出现了一个位于402.25 eV的峰,这与文献[29,30]中季铵化氮N1s结合能的数据一致,且在其I3d谱图中,在618.85 eV处出现一个新的特征峰,表明碘处于阴离子状态[31].氮原子特征区的新峰(402.25 eV)和I3d特征峰(618.85 eV)证明UiO-66-NH2已成功发生季铵化反应.
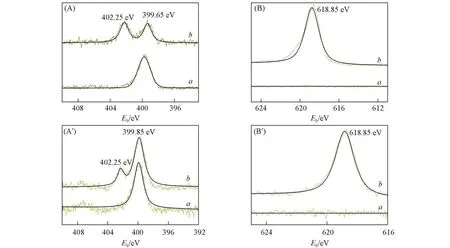
Fig.2 High-resolution XPS spectra for N1s(A,A′)and I3d(B,B′)of catalysts(A),(B)a.UiO-66-NH2;b.UiO-66-Ⅰ-3;(A′),(B′)a.Void@UiO-66-NH2;b.Void@UiO-66-Ⅰ.
为进一步确定催化剂的修饰程度,进行了1H NMR、ICP以及元素分析表征.1H NMR结果如图3所示,用圆点标记的3个特征峰的化学位移分别为δ7.78,δ7.38和δ7.06,可归属于未被修饰的配体苯环上的3个特征氢峰;用三角形标记的3个特征峰的化学位移分别为δ7.86,δ7.25和δ7.08,可归属于与等量的碘乙醇发生了烷基化反应的配体苯环上的3个特征氢峰;用菱形标记的3个特征峰的化学位移分别为δ8.39,δ8.21和δ8.10,归属于被季铵化成功的配体苯环上的3个特征氢峰.根据指定峰的积分,可以计算出样品的各成分含量.对于UiO-66-Ⅰ-1,有26%的氨基被季铵化成功,51%的氨基与等量的碘乙醇反应,还有23%的氨基未参加反应,将这一结果记为UiO-66-Ⅰ-1(季铵/仲胺/伯胺含量比为26∶51∶23),则其它样品的结果可分别记为UiO-66-Ⅰ-2(季铵/仲胺/伯胺含量比为37∶48∶15),UiO-66-Ⅰ-3(季铵/仲胺/伯胺含量比为47∶40∶13),Void@UiO-66-Ⅰ(季铵/仲胺/伯胺含量比为43∶43∶14).该结果表明随着反应次数的增加,参加烷基化反应的氨基数量增加,且季铵化程度越来越高.通过对UiO-66-NH2,UiO-66-Ⅰ和Void@UiO-66-Ⅰ进行元素分析和ICP测试(本文支持信息表S1),进一步证实了该结果.
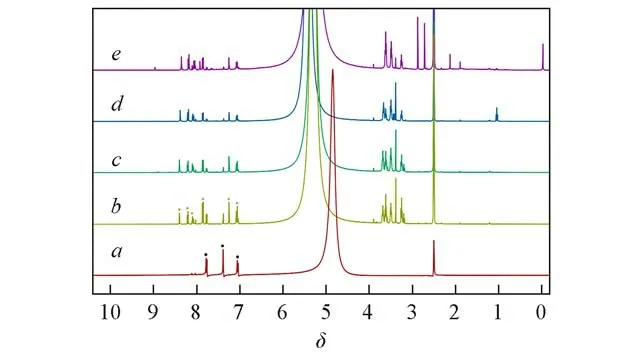
Fig.3 1H NMR spectra of digested UiO-66-NH2(a),UiO-66-I-1(b),UiO-66-I-2(c),UiO-66-I-3(d)and Void@UiO-66-I(e)Black dot:primary amine;blue triangle:secondary amine;red diamond:quaternary ammonium.
为了验证催化剂的形貌结构,对样品进行了宏观照片、SEM和TEM测试(图4).由图4(E)和(F)可见,对UiO-66-NH2进行后合成修饰后,样品颜色由淡黄色变为亮黄色,形貌为块状结构.图4(A)表明成功合成了表面光洁的SPS小球.在其表面生长UiO-66-NH2后,小球表面变粗糙,且样品颜色由白色变为淡黄色,表明成功合成SPS@UiO-66-NH2核壳结构[图4(B)].对SPS@UiO-66-NH2进行后合成修饰后,小球形貌无明显变化,样品颜色变为亮黄色[图4(C)和(D)].EDS能谱图[图4(I)~(L)]显示各元素均匀分布在MOFs骨架上,结合前面FTIR,XPS,1H NMR等测试结果,表明季铵盐被成功引入UiO-66中.由图4(G)和(H)可见,模板去除后小球基本没有破损且中间出现一个大空腔,表明中空结构构建成功.综合以上所有测试结果表明成功制备了空心纳米球Void@UiO-66-Ⅰ和块状UiO-66-Ⅰ这2种不同形貌的催化剂.

Fig.4 Photographs(insets)and SEM images of SPS(A),SPS@UiO-66-NH2(B),SPS@UiO-66-I(C),Void@UiO-66-I(D),UiO-66-NH2(E)and UiO-66-I(F),TEMimages of SPS@UiO-66-I(G)and Void@UiO-66-I(H),SEM-EDS mapping of Void@UiO-66-I[(I)—(L)](I)C;(J)Zr;(K)N;(L)I.
对UiO-66-NH2,UiO-66-Ⅰ,SPS@UiO-66-NH2,Void@UiO-66-NH2以及Void@UiO-66-Ⅰ在77 K下进行了氮气吸附-脱附表征来分析其孔道环境,结果如图5(A)、表S2和图S2(本文支持信息)所示.与未被修饰的UiO-66-NH2和Void@UiO-66-NH2相比,季铵化反应过后,UiO-66-Ⅰ和Void@UiO-66-Ⅰ比表面积均下降了约300 m2/g,且孔体积也相对减少,表明对UiO-66-NH2的后合成修饰取得成功,且生成的铵盐占据了MOFs的部分孔道,使得比表面积和孔容均显著降低.图5(B)为UiO-66-NH2,UiO-66-Ⅰ,Void@UiO-66-NH2以及Void@UiO-66-Ⅰ在273 K下测试得到的CO2吸附性能图,从图中可见,UiO-66-NH2和Void@UiO-66-NH2的CO2吸附量整体均比UiO-66-Ⅰ和Void@UiO-66-Ⅰ的高,且随着压强增加,它们之间的差值越大,这可能是因为氨基对CO2的吸附作用比季铵盐更好,引入季铵盐后占据了部分孔道或者阻碍了CO2与氨基接触,导致CO2吸附量下降.且在同类型材料中,中空结构的Void@UiO-66-NH2比实心结构的UiO-66-NH2的CO2吸附量高,表明中空结构有利于CO2吸附.
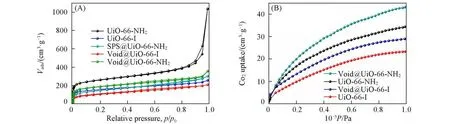
Fig.5 N2 adsorption-desorption isotherms at 77 K(A)and CO2 adsorption isotherms at 273 K(B)
为了研究材料的热稳定性,在流动氮气氛围下检测了材料的热失重数据(本文支持信息图S3).200℃之前的失重可能是由于在UiO-66孔道中物理吸附的水、DMF和残留乙酸的脱除而产生的.在UiO-66-Ⅰ,SPS@UiO-66-Ⅰ以及Void@UiO-66-Ⅰ的热重曲线中,于200℃左右出现了一个明显的失重,这与文献[32]报道的季铵盐热解温度一致,表明季铵化成功.在350℃之后出现的一个急剧失重则是由于UiO-66框架内的有机配体(对苯二甲酸)的分解[33].表明该材料具有良好的热稳定性,可以用于200℃以下催化CO2环加成反应.
2.2 催化剂的活性
成功合成并确认Void@UiO-66-Ⅰ结构后,认为将其用作CO2环加成反应制备环碳酸酯的多相催化剂具有以下优点:首先,其拥有稳定的UiO-66-NH2壳层;其次,引入MOFs内的季铵盐含有羟基和碘离子,羟基能与环氧化合物的氧原子形成氢键,碘离子能亲核攻击环氧化合物中位阻较小的碳原子,致使环氧化合物开环活化;最后,Void@UiO-66-Ⅰ催化剂内的大空腔不仅有利于吸附CO2,而且能加快传质速率,使反应更快地进行.以CO2与氧化苯乙烯的环加成反应为探针反应,对Void@UiO-66-Ⅰ空心纳米球的催化性能进行评价,并与实心块状形貌UiO-66-Ⅰ催化剂的催化活性进行比较,以证明Void@UiO-66-Ⅰ催化剂的结构优势,具体结果列于表1.

Table 1 Comparison of cycloaddition reaction of CO2 to styrene oxide over various catalysts a
从表1中可见,在反应温度120℃,CO2压力为1.2 MPa下,反应6 h时,UiO-66-NH2和Void@UiO-66-NH2催化CO2环加成反应生成碳酸苯乙烯酯的产率很低,分别只有9.0%和11.2%.以单独的四(2-羟基乙基)碘化铵为均相催化剂时,碳酸苯乙烯酯的产率可达94.7%,表明该季铵盐对二氧化碳和环氧化合物的环加成反应具有很好的催化作用.在其它反应条件不变的情况下,以Void@UiO-66-Ⅰ和UiO-66-Ⅰ作为催化剂,对比未引入季铵盐的催化剂,碳酸苯乙烯酯的产率显著提高,分别为85.5%和68.7%,这说明引入的季铵盐与UiO-66-NH2之间有良好的协同催化作用,能有效催化CO2和环氧化合物的环加成反应,且Void@UiO-66-Ⅰ的催化活性比UiO-66-Ⅰ的催化活性更好,表明中空结构能加快传质速率,从而提高催化活性.当改变反应条件,在反应温度为120℃,CO2压力为1.01×105Pa,反应时间为12 h时,Void@UiO-66-Ⅰ和UiO-66-Ⅰ仍然对CO2和氧化苯乙烯的环加成反应具有催化作用,且Void@UiO-66-Ⅰ表现出比UiO-66-Ⅰ更好的催化活性,进一步证明了中空结构比实心结构更有利于涉及气体的催化反应.
为了提高碳酸苯乙烯酯的产率,下面通过单因素实验探讨了Void@UiO-66-Ⅰ催化CO2和氧化苯乙烯反应生成碳酸苯乙烯酯的最佳反应条件,结果如图6所示.固定反应温度为120℃,CO2压力为1.2 MPa下,反应时间在2~8 h范围内,探讨反应时间对CO2环加成反应的影响,结果如图6(A)所示.随着反应时间的增加,碳酸苯乙烯酯的产率显著提高,反应时间为6 h时,产率最高,之后在很长一段时间内几乎保持不变.在反应温度为120℃,反应时间为6 h,CO2压力为0.8~1.4 MPa范围内探讨了CO2压强对CO2环加成反应的影响,结果如图6(B)所示.当CO2压强在0.8~1.2 MPa范围内逐步增加时,碳酸苯乙烯酯的产率也逐步增加,这可能是因为随着气相中CO2压强增加,在液相反应体系中的CO2浓度逐渐增加.当CO2压强进一步增加时,碳酸苯乙烯酯的产率不再快速增加,这可能是因为过高的CO2压强会减小在催化剂附近的环氧化合物的浓度,从而降低环氧化合物的转化率[34].之后在CO2压强为1.2 MPa、反应时间为6 h条件下,在反应温度为80~140℃范围内探讨了反应温度对CO2环加成反应的影响,结果如图6(C)所示.当反应温度在80~120℃范围内逐步增加时,碳酸苯乙烯酯的产率也逐步增加,但当反应温度为140℃时,底物转化率几乎不变,但选择性却降低了.根据以上研究结果,Void@UiO-66-Ⅰ为催化剂催化CO2与氧化苯乙烯发生环加成反应生成碳酸苯乙烯酯的最佳反应条件为反应温度为120℃,CO2压力为1.2 MPa,反应时间为6 h.
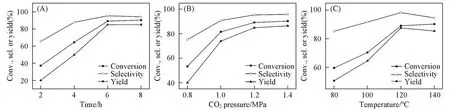
Fig.6 Catalytic activity of the Void@UiO-66-I for the cycloaddition reaction of CO2 and SO under different reaction time(A),CO2 pressure(B)and reaction temperature(C)

tions:25 mmol SO,0.5%(molar fraction)catalyst,120℃,1.2 MPa CO2,6 h;b.1.01×105 Pa CO2,12 h,all the other conditions are the same.
为了测试Void@UiO-66-Ⅰ催化剂的稳定性,对其进行滤出实验和循环实验,结果如图7所示.从图7(A)可见,去除催化剂后反应的转化率基本不变,表明反应时催化剂保持稳定.在上述最佳反应条件下进行循环实验,结果如图7(B)所示.催化剂循环5次后催化活性未见明显下降,且催化剂也几乎没有损失.对使用过后的催化剂进行表征(见本文支持信息图S4),发现其结构无明显变化,说明Void@UiO-66-Ⅰ催化剂具有良好的稳定性和可回收循环使用性.
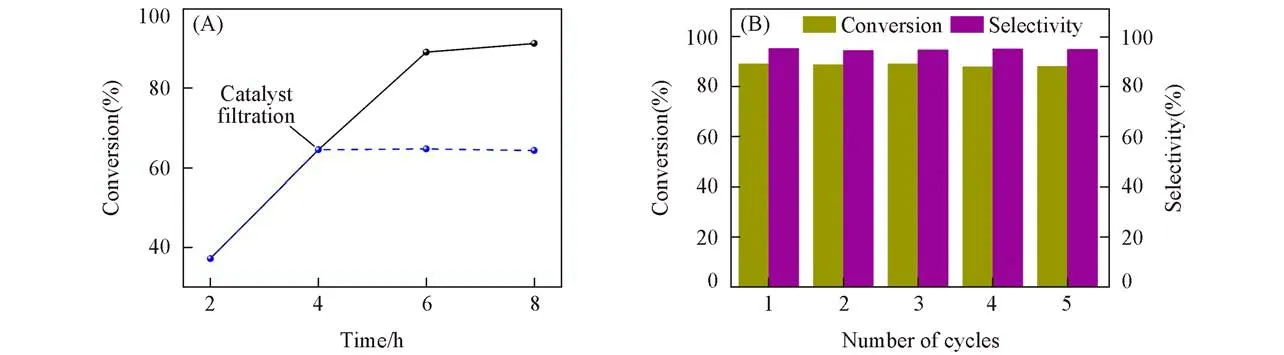
Fig.7 Filtration experiment(A)and reusability(B)of Void@UiO-66-I catalyst toward the cycloaddition reaction of CO2 and SO
结合文献[34,35]报道和本文实验数据,提出了Void@UiO-66-Ⅰ催化剂催化CO2环加成反应的可能机理,如Scheme 2所示.首先Void@UiO-66-Ⅰ中的羟基氢原子和氧化苯乙烯的氧原子之间形成氢键,导致环氧化合物C—O键的极化和中间体1的形成.然后卤素阴离子(I-)进攻氧化苯乙烯环氧环上空间位阻较小的碳,导致氧化苯乙烯开环和中间体2的形成.随后,CO2插入中间体2,从而形成烷基碳酸阴离子3.最后分子内发生亲核攻击导致分子内闭环,同时实现碳酸酯的形成和催化剂的再生.

Scheme 2 Plausible mechanism for the Void@UiO-66-I catalyzed cycloaddition of CO2 and SO
3 结 论
通过后合成修饰方法,合成了2种不同形貌的季铵盐功能化UiO-66-NH2(空心纳米球状Void@UiO-66-Ⅰ和实心块状UiO-66-Ⅰ),并通过一系列表征证明这2种催化剂的成功合成.依据催化剂的结构特点,将其应用于催化CO2和氧化苯乙烯环加成反应生成碳酸苯乙烯酯.在UiO-66-NH2中引入季铵盐后,催化活性显著提升,与实心块UiO-66-Ⅰ催化剂相比,中空纳米球Void@UiO-66-Ⅰ催化剂的催化活性得到进一步提高,其在最佳反应条件(120℃,1.2 MPa CO2和6 h)下,产物碳酸苯乙烯酯的产率和选择性分别为85.5%和95%,这归因于中空纳米球结构具有加速传质的特点.在中空结构的载体上引入高活性物种为构建高效的CO2环加成反应催化剂提供了参考.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20210221.
