铑催化吲哚与乙烯基三乙氧基硅烷的C—H烯基化反应
2021-08-16李鹏杰周春妮王泽田郑子昂张玉敏
李鹏杰,周春妮,王泽田,郑子昂,张玉敏,王 亮,肖 标
(江汉大学化学与环境工程学院,光电化学材料与器件教育部重点实验室,武汉 430056)
吲哚乙烯是许多天然产物和药物分子的核心结构单元[1],也是一种非常有用的合成中间体,可以用于构建咔唑及咔啉等吲哚类生物碱[2,3].尤其是末端吲哚乙烯,不仅在[4+2]环化反应、Heck偶联反应及烯烃交叉复分解反应中得到广泛应用,而且能够通过氧化或还原等反应转化为其它有价值的官能团[4].因此,吲哚乙烯衍生物的合成受到了化学家们的关注.近些年,过渡金属催化的C—H烯基化反应已成为制备这类化合物最有效的途径之一[5~9].相对于Heck,Suzuki和Still等传统偶联反应,C—H烯基化反应具有无需预先活化底物的优势[10].研究人员积极开展了相关研究工作,发展了各种烯基化偶联试剂.通过使用丙烯酸酯作为烯基化试剂,Wang等[11],Zhang等[12],Yi等[13]和Huang等[14]分别报道了吲哚的选择性C—H烯基化反应,通过改变吲哚氮原子上的导向基团,以控制吲哚2位C—H键的选择性活化.Carretero等[15]和Glorius等[16]分别研究了苯乙烯作为烯基化试剂参与吲哚的C—H烯基化反应,但苯乙烯底物的适用范围很窄.Li等[17]和Loh等[18]分别研究了2,2-二氟苯乙烯与吲哚的C—H烯基化反应,底物适用范围非常广泛,为制备含氟吲哚乙烯提供了新方法.Ackermann等[19]首次报道了CoI2催化的N-嘧啶吲哚与烯醇酯类化合物的C—H烯基化反应,该反应对于环状的烯醇酯具有较好的效果,非环状的烯醇酯收率则较低.尽管吲哚的C—H烯基化反应已经取得了一些进展,但这些研究成果主要用于制备非末端吲哚乙烯衍生物,对于末端吲哚乙烯的制备无效.2014年,Li等[20]使用α-取代的丙烯酸作为烯基化试剂,实现了一种[Rh(COD)2]OTf催化吲哚的C—H烯基化反应,可以合成末端吲哚乙烯,但该反应底物范围很窄,仅有4个实例.2019年,Punji等[21]使用α-溴烯烃衍生物作为烯基化试剂,通过Ni(bipy)Br2催化N-吡啶吲哚的C—H烯基化反应实现了末端吲哚乙烯的合成,但该反应需要使用强碱t-BuOK,且底物范围很窄,收率不高.最近,本课题组[22]使用乙烯基三氟硼酸钾作为烯基化试剂,温和反应条件下实现了二氯(五甲基环戊二烯基)合铑(Ⅲ)二聚体{[RhCp*Cl2]2(Cp*:五甲基环戊二烯基)}催化的N-嘧啶吲哚C—H烯基化反应,制备了各种官能化的末端吲哚乙烯.
基于前文[23~26]对C—H键活化反应的研究成果,本文使用商业化购买的乙烯基三乙氧基硅烷作为烯基化试剂,研究了[RhCp*Cl2]2催化N-嘧啶吲哚与乙烯基三乙氧基硅烷的C—H烯基化反应,进一步丰富了末端吲哚乙烯衍生物的制备方法.
1 实验部分
1.1 试剂与仪器
乙烯基三乙氧基硅烷、氟化银(AgF)、四氟硼酸银、二氟氢化钾、氟化锂、四丁基氟化铵(TBAF)和乙酸铜[Cu(OAc)2]购自萨恩化学技术(上海)有限公司;乙醇钠(NaOEt)、氢化钠(NaH)、溴化苄(BnBr)、甲氧基胺盐酸盐(H2NOMe·HCl)和无水硫酸镁购自上海贤鼎生物科技有限公司;所用试剂均为市售分析纯产品,未经过纯化直接使用.乙腈、N,N-二甲基甲酰胺(DMF)、甲苯、二甲亚砜(DMSO)、1,2-二氯乙烷(DCE)、四氢呋喃(THF)、叔丁醇(t-BuOH)、1,4-二氧六环(1,4-Dioxane)和乙酸乙酯等均为分析纯,购自国药集团化学试剂有限公司;[RhCp*Cl2]2购于上海贤鼎生物科技有限公司;所用溶剂均按照《实验室化学品纯化手册》[27]进行干燥处理;GF254薄层层析和柱层析硅胶(200~300目,山东青岛海洋化工厂生产).
Bruker AvanceⅡ-400 MHz型超导核磁共振波谱仪(NMR,德国Bruker公司);Bruker micrOTOF-QII型高分辨质谱仪(HRMS,德国Bruker公司);WRS-2型微机熔点仪(上海圣科仪器设备有限公司).
1.2 实验过程
C—H烯基化反应的合成路线如Scheme 1所示.在氩气保护下,向干燥的10 mL反应管中依次加入0.2 mmolN-嘧啶吲哚(1a)、0.6 mmol乙烯基三乙氧基硅烷(2)、0.4 mmol Cu(OAc)2、0.4 mmol AgF、0.004 mmol[RhCp*Cl2]2和2 mL DCE;将反应混合物于90℃反应24 h;冷却至室温,加入20 mL乙酸乙酯稀释后过滤并浓缩;粗产品经硅胶柱层析纯化[V(乙酸乙酯)∶V(石油醚)=1∶20~1∶10]得化合物3a.采用相同方法合成化合物3b~3z.

Scheme 1 C—H alenylation of indoles and vinyltriethoxysilane
Scheme 2给出2-乙烯基吲哚-3-甲醛(4)的合成路线.向干燥的25 mL反应瓶中依次加入1.0 mmol 2-乙烯基-N-嘧啶吲哚-3-甲醛(3t)、4.0 mmol NaOEt和8 mL DMSO;将反应混合物于110℃反应15 min;冷却至室温,加入冰水(20 mL)淬灭;用乙酸乙酯(20 mL×3)萃取,用20 mL饱和NaCl溶液洗涤2次,无水硫酸镁干燥后过滤并浓缩;粗产品经硅胶柱层析纯化[V(乙酸乙酯)∶V(石油醚)=1∶15~1∶10]得到化合物4.

Scheme 2 Synthetic routes ofδ-carboline
Scheme 2给出2-乙烯基-N-苄基吲哚-3-甲醛(5)的合成路线.在氩气保护下,向干燥的10 mL反应瓶中依次加入0.4 mmol化合物4、4 mL DMF,将反应体系冷却至0℃;分批加入0.44 mmol NaH反应30 min;再分批加入0.48 mmol溴化苄,然后升至室温反应3 h;加入5 mL冰水淬灭反应,用乙酸乙酯(20 mL×3)萃取,用20 mL饱和NaCl溶液洗涤2次,无水硫酸镁干燥后过滤并浓缩;粗产品经硅胶柱层析纯化[V(乙酸乙酯)∶V(石油醚)=1∶10~1∶5],得到化合物5.
Scheme 2给出δ-苄啉衍生物(6)的合成路线.向干燥的10 mL反应管中依次加入0.2 mmol化合物5、0.4 mmol NaOAc、0.4 mmol NH2OH·HCl和2 mL 1,4-二氧六环;将反应混合物于110℃回流反应24 h;冷却至室温,加入10 mL二氯甲烷稀释后浓缩;粗产品经硅胶柱层析纯化[V(乙酸乙酯)∶V(石油醚)=1∶5~1∶2],得到化合物6.
表1给出化合物3a~3z和4~6的理化数据,核磁共振波谱数据见表2和3,核磁共振谱图见图S1~S37(见本文支持信息).

Table 1 Appearance,melting points and HRMS data of compounds 3a—3z and 4—6

Table 2 1H NMR and 13C NMR data of compounds 3a—3u and 3x

Continued
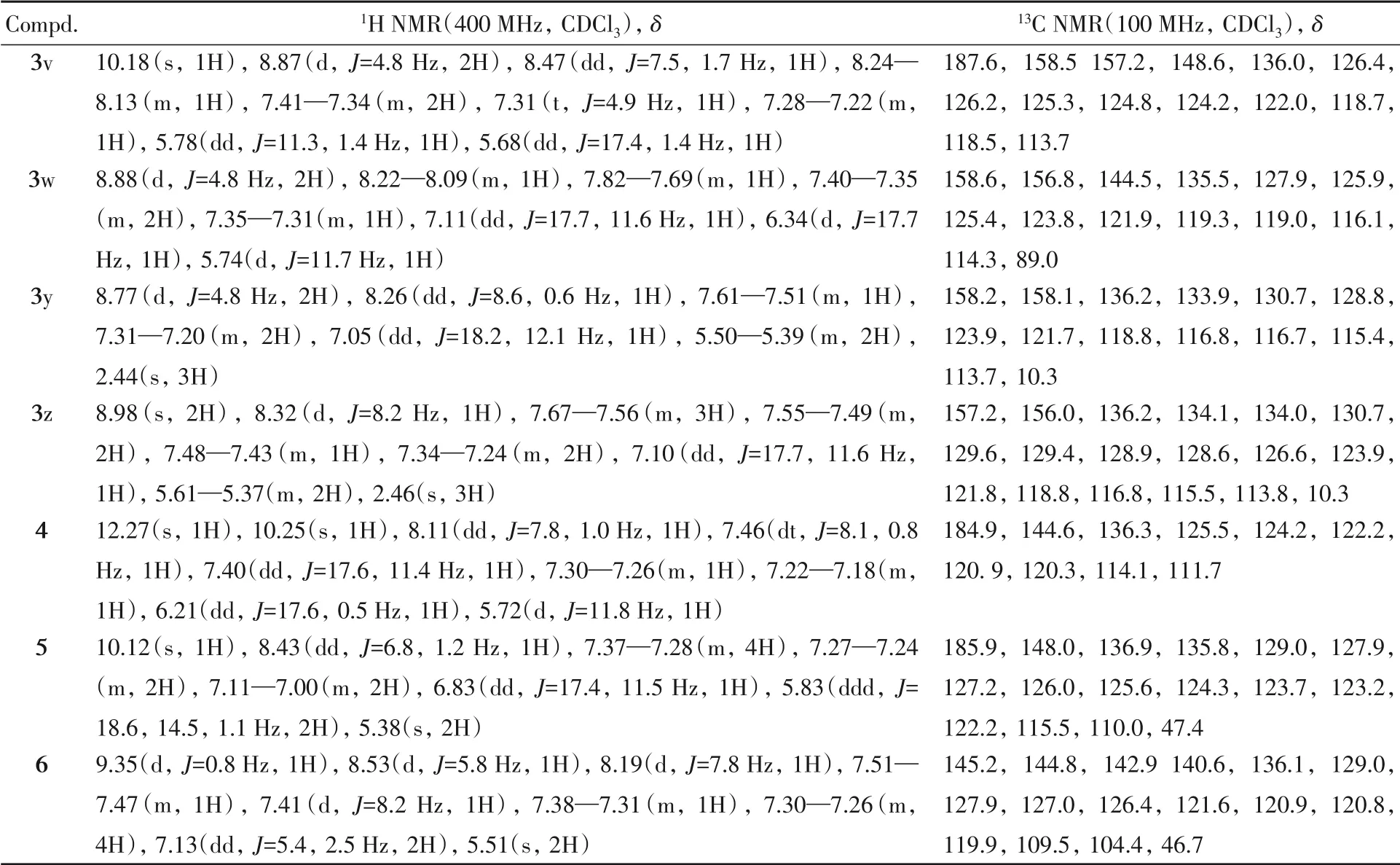
Table 3 1H NMR and 13C NMR data of compounds 3v,3w,3y,3z and 4—6
2 结果与讨论
2.1 反应条件的优化
以化合物1a作为模型底物,化合物2作为烯基化试剂,化合物2与1a的摩尔比为3.0∶1,[RhCp*Cl2]2作为催化剂,乙酸酮作为氧化剂,氟化银作为添加剂,反应温度为100℃,对溶剂进行了筛选,结果列于表4.可见,当使用乙腈或DMF作为溶剂时,收率为51%(表4中Etries 1和2);以甲苯作为溶剂时,收率为42%(表4中Entry 3);使用DMSO时,收率提高至70%(表4中Entry 4);以1,2-二氯乙烷作为溶剂时,收率进一步提高到72%(表4中Entry 5).尝试用四氢呋喃、叔丁醇及1,4-二氧六环等溶剂,反应均不能发生(表4中Entries 6~8);使用1,2-二氯乙烷和DMF混合溶剂(体积比为1∶1)时,收率为65%(表4中Entry 9).在此基础之上,对反应温度进行了考察.当温度升至110℃时,收率略有上升(表4中Entry 10);继续升温至120℃,收率下降至55%(表4中Entry 11).当降温至90℃时,收率提高到88%(表4中Entry 12);继续降低温度至80℃,收率下降至43%(表4中Entry 13);含氟添加剂的优化结果表明,使用四氟硼酸银时,收率为67%(表4中Entry 14);使用二氟氢化钾时,收率为60%(表4中Entry 15);使用氟化锂或四丁基氟化胺时,反应不能发生(表4中Entries 16和17);未使用含氟添加剂时,反应也不能发生(表4中Entry 18).最后,对乙烯基三乙氧基硅烷(2)的用量进行了考察.结果表明,结果表明,降低化合物2与1a的摩尔比至1∶1和1.5∶1时,有少量原料未反应完全,收率均为82%;当二者摩尔比为2∶1时,仍有少量原料未反应完全,收率为74%;当二者摩尔比为2.5∶1时,原料基本反应完全,收率为85%;增加二者摩尔比至3.5∶1时,收率明显下降;当摩尔比为4∶1时,收率仅有52%.综上所述,最佳的反应条件是使用[RhCp*Cl2]2作为催化剂,Cu(OAc)2为氧化剂,AgF为添加剂,1,2-二氯乙烷为溶剂,反应温度为90℃,化合物2与1a的摩尔比为3.0∶1.
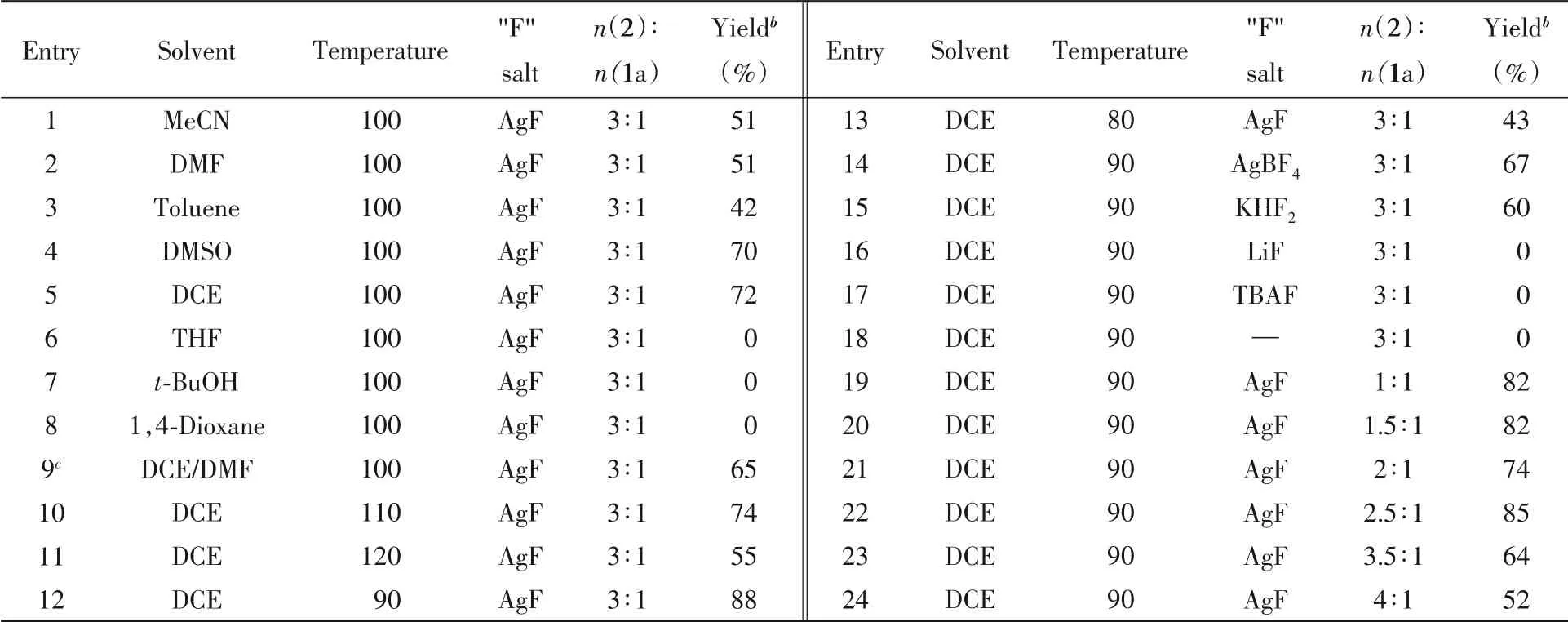
Table 4 Optimization of the reaction conditions a
2.2 底物的适用范围
在最佳反应条件下,考察了反应的底物适用性,结果列于表5.可见,在吲哚环上引入各种供电子基团,如甲基、甲氧基和苄氧基时,均能较好地适应反应体系,收率为60%~71%(表5中Entries 2,4,5,10和14).实验过程中发现,这些底物可以完全转化,但反应中存在底物分解现象.在吲哚环上引入各种吸电子基团,如氟、氯、溴、氰基和硝基时,反应也能够顺利地进行,收率为53%~79%(表5中Entries 3,6~9和11~13).其中,卤素基团为官能团的转化提供了反应位点.对吲哚碳3位取代基的适用范围进行了考察,各种取代基都能够兼容,如乙基、丁基、异丙基、烯丙基、苄酯基和乙酰氧基等,收率为38%~65%(表5中Entries 15~21).具有醛基和氰基的N-嘧啶吲哚也适用于反应体系(表5中Entries 22和23),由于原料没有反应完全,因此收率仅有42%和44%.此外,嘧啶环上具有溴、甲基或苯基取代的吲哚,也是较好的反应底物,收率为47%~72%(表5中Entries 24~26).

Table 5 Substrate scope of C—H alkenylation of indoles and vinyltriethoxysilane
2.3 同位素效应实验
在最佳反应条件下进行了同位素效应实验.将N-嘧啶吲哚(1a)与氘代的N-嘧啶吲哚(d-1a)等量混合,与化合物2在同一反应体系中反应6 h.采用核磁共振波谱表征未反应的原料,结果表明,化合物1a与d-1a的摩尔比为5.7∶1.因此,推断该反应的动力学同位素效应KH/KD=5.7∶1,推测C—H键的断裂可能是反应过程中的决速步骤.
2.4 竞争性实验
在最佳反应条件下,将3,5-二甲基-N-嘧啶吲哚(1d)和3-甲基-5-硝基-N-嘧啶吲哚(1i)于同一个反应体系中进行竞争性实验,产物3d和3i的分离收率分别为45%和25%.结果表明,反应中含有给电子基团的底物比吸电子基团的底物具有更高的反应活性.因此,推断C—H键断裂方式可能是一种碱协助内部亲电性取代过程[28].
2.5 反应机理
本文提出的反应机理如Scheme 3所示.以化合物1a和2的反应为例:首先,催化剂[RhCp*Cl2]2被Cu(OAc)2活化,形成具有亲电性的催化物种Cp*RhOAc2.随后,Cp*RhOAc2与化合物1a的氮原子发生配位生成中间体B.通过亲电性金属化以及乙酸根负离子去质子化过程断裂邻位C—H键,中间体B转化为五元环状铑络合物C,该步骤是反应过程中的决速步骤.化合物2被氟离子活化后产生五价的硅烷中间体F[29].然后,中间体F与铑络合物C发生转金属化反应,形成铑络合物D.经过还原消除反应形成产物3a,并释放出Cp*Rh(I).最后,Cp*Rh(I)被Cu(II)或者Ag(I)氧化为Cp*Rh(OAc)2,完成整个催化循环.

Scheme 3 Proposed mechanism of the reaction
2.6 合成应用
咔啉是一类广泛分布于自然界的含氮生物碱,具有抗菌、抗病毒、抗肿瘤及中枢神经系统抑制等活性.将本文反应应用于δ-苄啉衍生物的制备,如Scheme 2所示,产物3t移除嘧啶基团后,以42%的收率获得2-乙烯基吲哚-3-甲醛(4).经过苄基化反应,化合物4能够转化为产物5,收率为85%.最后,化合物5与甲氧基胺盐酸盐发生环化反应获得δ-咔啉衍生物(6),收率为73%.
3 结 论
使用商业化购买的乙烯基三乙氧基硅烷作为烯基化试剂,实现了[RhCp*Cl2]2催化N-嘧啶吲哚与乙烯基三乙氧基硅烷的直接C—H烯基化反应.考察了溶剂、温度、添加剂对合成末端吲哚乙烯衍生物的影响,在最佳反应条件下,以42%~88%的收率得到了末端吲哚乙烯类化合物.该方法具有良好的底物普适性,含有给电子取代基和吸电子取代基的N-嘧啶吲哚均能顺利发生转化.通过核磁共振波谱表征推算动力学同位素效应KH/KD=5.7∶1.因此,推断C—H键断裂可能是反应过程中的决速步骤.研究了底物的竞争性实验,结果表明,给电子取代基的底物具有更高的反应活性.因此,推断C—H键断裂方式可能是一种碱协助内部亲电性取代过程.最后,将该合成方法成功应用于一种δ-咔啉衍生物的制备.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20210107.
