Genome and population evolution and environmental adaptation of Glyptosternon maculatum on theQinghai-Tibet Plateau
2021-08-16ShiJunXiaoZenBoMouRuiBinYangDingDingFanJiaQiLiuYuZouShiLinZhuMingZouChaoWeiZhouHaiPingLiu
Shi-Jun Xiao ,Zen-Bo Mou ,Rui-Bin Yang ,Ding-Ding Fan ,Jia-Qi Liu ,Yu Zou ,Shi-Lin Zhu,Ming Zou,Chao-Wei Zhou,Hai-Ping Liu
1 Institute of Fisheries Science, Tibet Academy of Agricultural and Animal Husbandry Sciences, Lhasa, Tibet 810000, China
2 Department of Computer Science, Wuhan University of Technology, Wuhan, Hubei 430070, China
3 College of Fisheries, Huazhong Agricultural University, Wuhan, Hubei 430070, China
4 Key Laboratory of Freshwater Fish Reproduction and Development (Ministry of Education), College of Fisheries, Southwest University,Chongqing 402400, China
5 College of Plant Protection, Jilin Agriculture University, Changchun, Jilin 130118, China
6 Jiaxing Key Laboratory for New Germplasm Breeding of Economic Mycology, Jiaxing, Zhejiang 314000, China
ABSTRACT Persistent uplift means the Qinghai-Tibet Plateau(QTP) is an ideal natural laboratory to investigate genome evolution and adaptation within highland environments.However,how paleogeographic and paleoclimatic events influence the genome and population of endemic fish species remains unclear.Glyptosternon maculatum is an ancient endemic fish found on the QTP and the only critically endangered species in the Sisoridae family.Here,we found that major transposons in the G.maculatum genome showed episodic bursts,consistent with contemporaneous geological and climatic events during the QTP formation.Notably,histone genes showed significant expansion in the G.maculatum genome,which may be mediated by long interspersed nuclear elements (LINE) repetitive element duplications.Population analysis showed that ancestral G. maculatum populations experienced two significant depressions 2.6 million years ago (Mya) and 10 000 years ago,exhibiting excellent synchronization with Quaternary glaciation and the Younger Dryas,respectively.Thus,we propose that paleogeography and paleoclimate were dominating driving forces for population dynamics in endemic fish on the QTP.Tectonic movements and temperature fluctuation likely destroyed the habitat and disrupted the drainage connectivity among populations.These factors may have caused severe bottlenecks and limited migration among ancestral G.maculatum populations,resulting in the low genetic diversity and endangered status of the species today.
Keywords: Qinghai-Tibet Plateau (QTP);Glyptosternon maculatum; Genome evolution;Population; High-altitude adaptation
INTRODUCTION
Deciphering how genomes evolve in response to dramatic environmental change is an essential question in evolutionary biology to understand the molecular mechanisms of adaptations and speciation.The Qinghai-Tibet Plateau (QTP)was formed from the collision of the Indian and Eurasian Plates and is the youngest,largest,and highest plateau on Earth,with an average altitude of over 4 000 m a.s.l.(Peng et al.,2006).Continued uplift over the last 50 million years ago(Mya) (Peng et al.,2006) and the extreme climatic environment make the QTP an ideal and unique natural laboratory to investigate how paleogeography and paleoclimate have influenced endemic genome and population evolution.The high-altitude environment of the QTP is characterized by high ultraviolet (UV) radiation,dramatic temperature changes,and nutritional deficiencies (Jiang et al.,2012;Zhang et al.,2016).Consequently,understanding genome and population evolution is essential to gain insight into the molecular mechanisms underlying environmental adaptations and to protect the wild genetic resources of endemic endangered species in the QTP.
Research has elucidated the molecular mechanisms that underlie the high-altitude adaptations of Tibetan people(Petousi & Robbins,2014;Yi et al.,2010).However,the initial peopling of the QTP by modern humans occurred about 25 000 years ago (Beall,2007),which is significantly more recent than the long history of several million years for QTP formation.Endemic animals of the plateau represent excellent genome evolution models with long adaptive histories during plateau uplift.In the last decade,studies on genome sequencing in Tibetan animals (Wang et al.,2014),including yaks (Qiu et al.,2012),pigs (Ai et al.,2014),dogs (Gou et al.,2014),antelopes (Ge et al.,2013),and birds (Qu et al.,2013),have broadened our understanding of genomic adaptations to the local environment via the evolution and selection of putatively important functional genes (Jablonski,2017;Wei et al.,2016).However,few studies have uncovered genome characterizations of endemic animals. Moreover,the relationship between the evolution of endemic species genomes and populations and paleogeographic and paleoclimatic events during the formation of the QTP is still largely unknown.
The dispersal and distribution of fish strictly depend on drainage connections,which are highly influenced by tectonic movements and climate change (Yang et al.,2009).Thus,the genomic evolution of freshwater fish offers an excellent opportunity to contextualize biological evolution to changes in QTP geology and climate (Kang et al.,2017;Xiao et al.,2020;Yang et al.,2009).Although transcriptomic and genomic studies onSchizothorax o'connori(Xiao et al.,2020) andTriplophysa bleekeri(Yuan et al.,2020) have revealed differential expression and natural selection of key genes in QTP fish adaptations (Ma et al.,2016),research on fish genome and population evolution in relation to environmental change and uplift of the QTP remains limited.The family Sisoridae in the order Siluriformes,which contains glyptosternoid and non-glyptosternoid species,is one of the main taxonomic groups of fish in the basin waters of the QTP(Peng et al.,2006).The evolution and speciation of sisorid catfish are thought to have been profoundly influenced by QTP uplift (Guo et al.,2005;He et al.,2001).Glyptosternon maculatumis one of the most ancient species within Glyptosterninae and is distributed at altitudes from 2 800–4 200 m in the Yarlung Zangbo River (Zhang et al.,2010).Consequently,due to its peculiar ecological distribution and evolutionary history (Zhang et al.,2011),G.maculatumis an ideal species to explore genome and population evolution during the formation of the QTP and to evaluate how genomic changes contributed to the environmental adaptations of sisorid catfish on the QTP.
To this end,we performedG.maculatumgenomic analysis and revealed episodic transposon bursts,which may have facilitated functional gene expansions and thus contributed to the speciation and environmental adaptation ofG.maculatum.Population genetic analyses based on whole-genome variants were performed to address how contemporaneous environmental changes may have influenced ancestral populations.We revealed that both QTP uplift and climatic events likely imposed immense natural stress onG.maculatumand limited migration among populations,leading to the low genetic diversity and endangered status ofG.maculatumtoday.
MATERIALS AND METHODS
Chromosomal construction and transcriptome assembly for phylogenetic analysis
We used Hi-C techniques for chromosomal assembly ofG.maculatumbased on contig sequences assembled from a previous study (Edgar,2004).Muscle samples from the sameG.maculatumfish used for genome sequencing were used for Hi-C library construction following prior research (Gong et al.,2018).The Hi-C libraries were controlled for quality and sequenced on the Illumina Hiseq X Ten platform (Illumina,USA) (Supplementary Table S1).The contig sequences ofG.maculatumwere scaffolded into chromosomes using the bioinformatics method in Gong et al.(2018) (Supplementary Table S2 and Figure S1).
Transcriptomes of muscle tissue from 16 QTP catfish(Supplementary Tables S3,S4),including 15 Sisoridae fish(Exostoma labiatum,Pareuchiloglanis feae,Pareuchiloglanis kamengensis,Glyptothorax quadriocellatus,Glyptothorax fukiensishonghensis,Glyptothoraxinterspinalum,Glyptothorax cavia,Glyptothorax dorsalis,Glyptothorax laosensis,Glyptothorax zainaensis,Glyptothorax trilineatus,Glyptothorax minimaculatus,Bagarius yarrelli,andPseudecheneis sulcatus) and one Bagridae fish (Leiocassis longirostris),were sequenced using the Illumina HiSeq X Ten platform (Illumina,USA).In total,66 Gb of raw RNA-seq data were generated.The RNA-seq reads of these fish were assembled by Trinity v2.8.6 (Kumar et al.,2016)(Supplementary Table S4).
Phylogenetic analysis among sisorid catfish
The unigenes of the 16 catfish were translated into protein sequences.Protein sequences longer than 50 amino acids were clustered with the whole proteome ofG.maculatumusing OrthoMCL v2.0.9 (Li,2011).We obtained 167 singlecopy genes with a total length of~232 kb after gene family clustering.MUSCLE v3.8.31 (Li & Durbin,2009) was used to generate multiple sequence alignments for protein sequences in each single-copy family with default parameters.The alignments of each family were concatenated to a super alignment matrix,which was used for phylogenetic tree reconstruction with RAxML v8 (Li et al.,2003).Divergence time between fish species was estimated using MCMCTree in Phylogenetic Analysis by Maximum Likelihood (PAML) v4(Lieberman-Aiden et al.,2009),with the options “correlated molecular clock” and “JC69” model.Markov Chain Monte Carlo analysis was run for 20 000 generations using a burn-in of 1 000 iterations.Four calibration points were applied:GlyptosternonandExostomadivergence time (5.5–8.8 Mya),GlyptosternonandGlyptothoraxdivergence time (7.7–12.2 Mya),GlyptosternonandPseudecheneisdivergence time(7.7–12.2 Mya),and Sisoridae and Bagridae divergence time(41–143 Mya).These calibration points were traced from TimeTree (http://www.timetree.org/) (Supplementary Figure S2).
Expanded gene families and positively selected gene detection
The phylogenetic tree and divergence times ofG.maculatumand other species were analyzed using CAFE v5 (De Bie et al.,2006) to infer changes in gene family size using a probabilistic model.Gene Ontology (GO) enrichment analysis was performed on theG.maculatumexpanded genes using the topGO v2.36.0 package (Alexa et al.,2006).MUSCLE v3.8.31 was used for multi-protein sequence alignment of theG.maculatumgenes and their orthologs.Conserved CDS alignments of each single-copy gene family were extracted by Gblocks v0.91b (Talavera & Castresana,2007) and used for further identification of positively selected genes.The ratios of nonsynonymous to synonymous substitutions (KA/KS,or ω)were estimated for each single-copy orthologous gene using the codeml program with the branch-site model implemented in PAML v4 (Guindon et al.,2010).A likelihood ratio test was conducted,and false discovery rate (FDR) correction was performed for multiple comparisons.Genes with correctedP<0.01 were defined as naturally selected genes.
Phylogeny and burst time estimation of LINE RTE-BovB and L2 type
Repetitive sequences in theG.maculatumgenome were identified by a combination of homology searching andab initioprediction.For homology-based prediction,we used RepeatMasker v4.1.2 (Bergman & Quesneville,2007) and ProteinMask to search against RepBase.Forab initioprediction,we used Tandem Repeats Finder (TRF) v4.04(Benson,1999),LTR_FINDER v1.0 (Xu & Wang,2007),PILER v1.3.11 (Edgar & Myers,2005),and RepeatScout v1.0.5 (Price et al.,2005) with default parameters.We found that at least 32% of theG.maculatumassembly was composed of repetitive elements (Supplementary Table S5).The identified repeats were annotated using RepBase v23.04(Jurka et al.,2005) with RepeatMasker v4.1.2 (Bergman & Quesneville,2007) against this database.Other species,includingAstyanax mexicanus,Cyprinus carpio,Danio rerio,Ictalurus punctatus,Oncorhynchus mykiss,Oryzias latipes,Pelteobagrus fulvidraco,andPoecilia formosa,were also detected using the same pipeline (Supplementary Tables S5,S6 and Figures S3,S4).Interestingly,we found that long interspersed nuclear elements (LINE) content was much more abundant in theG.maculatumgenome.
The times of transposon bursts were estimated using previously published methods (Albertin et al.,2015).We dated transposable elements longer than 500 bp with RepeatMasker and adjusted the distances for multiple substitutions using the Jukes-Cantor formula:

where d is the distance estimated by RepeatMasker v4.1.2.Using an estimate of 0.003 6 JK/myr for synonymous substitutions per million years (Albertin et al.,2015;Xu et al.,2014),the divergence time of the repeats was then estimated.
Population genetic analysis for G.maculatum using whole-genome data
Ten living individuals were collected from 2900,4100-1,and 4100-2 using gill nets.All individuals were narcotized with MS-222 (Solarbio,China) for a few minutes before sampling muscles.For each individual,the muscle samples were immediately frozen in liquid nitrogen after dissection and stored at −80 °C until DNA extraction.Muscle samples from the outgroup,E.labiatum,were also collected.
For each sample,genomic DNA was extracted and used for library construction.Sequencing libraries were generated using a Truseq Nano DNA HT Sample Preparation Kit(Illumina,USA) following the manufacturer’s recommendations.Fragments with a length of 350 bp were selected,end polished,A-tailed,and ligated with the full-length adapter for Illumina sequencing.After polymerase chain reaction (PCR) amplification,all products were purified(AMPure XP system) and analyzed for size distribution using an Agilent2100 Bioanalyzer (Agilent Technologies,USA) and quantified using real-time PCR.Sequencing was implemented by the Illumina Hiseq 2000 platform (Illumina,USA) and millions of 150 bp paired-end short reads were generated.
Raw data were appraised using FastQC v.0.10.1 and filtered using fastp v0.20.0 (Chen et al.,2018).The remaining high-quality paired-end reads were mapped to the genome using BWA6 v0.7.8 (Li & Durbin,2010) with the command“mem -t 4 -k 32–M”.To reduce mismatches generated by PCR amplification before sequencing,duplicate reads were removed by SAMtools v1.9 (Li,2011).Single nucleotide polymorphism (SNP) calling was performed following the Genome Analysis Toolkit (GATK) v4.1.9.0 best practices(McKenna et al.,2010).Annotations were performed using the ANNOVAR package (v2013-05-20) (Wang et al.,2010).
Based on the identified SNPs,a neighbor-joining tree representing the relationships of each individual was constructed using TreeBeST v1.9.2 (http://treesoft.sourceforge.net/treebest.shtml) with 100 bootstrap resamplings.Based on the same data,the population genetic structure was estimated using the Bayesian computer algorithm with the R package LEA v3.13 (Frichot & François,2015). Principal component analysis (PCA) was also implemented using R v4.1.0.
Demographic history of G.maculatum
The population history of three populations ofG.maculatumwas constructed using the multiple sequentially Markovian coalescent (MSMC) approach v1.1.0 (Schiffels & Durbin,2014) with a generation time of 6 and mutation rate of 3.51×10−9per year per nucleotide (Yang et al.,2016).MSMC uses a hidden Markov model to analyze patterns of heterozygosity along genome sequences.We also applied the Generalized Phylogenetic Coalescent Sampler (G-PhoCS)v1.2.3 (Gronau et al.,2011).The prior distributions over model parameters were defined by Gamma distributions (Gronau et al.,2011).Markov chain was run for 100 000 iterations and population parameter values were sampled every 10 iterations.
RESULTS
Genome and transcriptome assembly for phylogenetic analysis of sisorid catfish
Based on the contigs assembled from the PacBio long reads(Liu et al.,2018),Hi-C sequencing was applied with the sameG.maculatumsample collected in the Tibet Plateau to obtain a high-quality chromosomal genome.A 704.8 Mb genome was obtained with a contig and scaffold N50 of 878.4 kb and 27.8 Mb,respectively.More than 91.7% of the sequences were anchored on chromosomes at the base level,resulting into 2 083 genomic sequences longer than 2 kb(Supplementary Table S2 and Figure S1).The GC content of theG.maculatumgenome was 39.6%.The transcriptomes of 16 sisorid catfish andLeiocassis longirostriswere sequenced and assembled for the following analyses (Supplementary Table S3).
The phylogenetic relationships ofG.maculatumto the other 16 sisorid catfish species were constructed using the transcriptome data (Figure 1;Supplementary Table S3),withL.longirostrisused as the outgroup species (Figure 2A).Pseudecheneiswas the earlier diverging genus among Sisoridae,and glyptosternoid and non-glyptosternoid fish clearly formed two sister groups.GlyptosternonandExostomawere primitive taxa among the glyptosternoid fish,whileBagariuswas primitive in the non-glyptosternoid group.ThePseudecheneisgenus diverged from the common ancestor of Sisoridae~9.7 Mya and glyptosternoid and non-glyptosternoid fish separated~8.1 Mya.
Protein-coding genes were used to reconstruct a genomebased phylogeny forG. maculatumamong teleosts(Figure 2B).The analysis supported a close evolutionary relationship betweenI.punetausandP.fulvidraco,consistent with the traditional taxonomy of Siluriformes.From our phylogenetic analysis,G.maculatumdiverged from their common ancestor withI.punetausandP.fulvidraco37–43 Mya (Supplementary Figure S2),a time that roughly corresponds to the collision of the ancient Indian and Eurasian plates in the Cenozoic era (Li et al.,2015).

Figure 1 Sampling sites (red spot) for fish species on the Qinghai-Tibet Plateau
Repetitive elements in G.maculatum genome
More than 30% of theG.maculatumgenome was composed of repetitive sequences (Supplementary Table S5).The abundance of whole genomic repeats in theG.maculatumgenome was comparable to that of other teleost fish,but the abundance of long interspersed elements (LINEs) comprised>23.1% and >66.3% of the genome and repetitive elements,respectively,significantly higher than that observed in other teleost fish (Figure 2B;Supplementary Table S4).LINEs(primarily LINE/L1 types) comprise~18.9% of the human genome (Li et al.,2001),but do not dominate repeat regions in other teleost genomes (Supplementary Table S6).The two most abundant LINE transposons in theG.maculatumgenome were RTE-BovB (93.4 Mb long and 12.3% of the genome) and L2 (57.9 Mb and 7.6%) (Supplementary Table S6).The abundance of long terminal repeats (LTRs) was also slightly higher in theG.maculatumgenome relative to the other teleosts,but DNA transposon abundance was lower(Supplementary Table S6).
Timing of LINE expansions in G.maculatum genome
The timing of LINE transposon expansions was estimated from sequence comparisons of LINE RTE-BovB and L2.The estimated time for expansion bursts indicated a multi-stage expansion pattern for both types (Figure3;Supplementary Figures S3,S4).The first stage of expansion occurred slowly during the late Eocene and early Oligocene~30 Mya.The second stage exhibited an even more substantial expansion during the early Miocene~20 Mya.The third stage showed a sharp expansion starting from 3 Mya in the Pleistocene.We observed remarkable associations of tectonic movements and global surface temperature fluctuations to the historical RTEBovB and L2 bursts (Figure 3A,B).
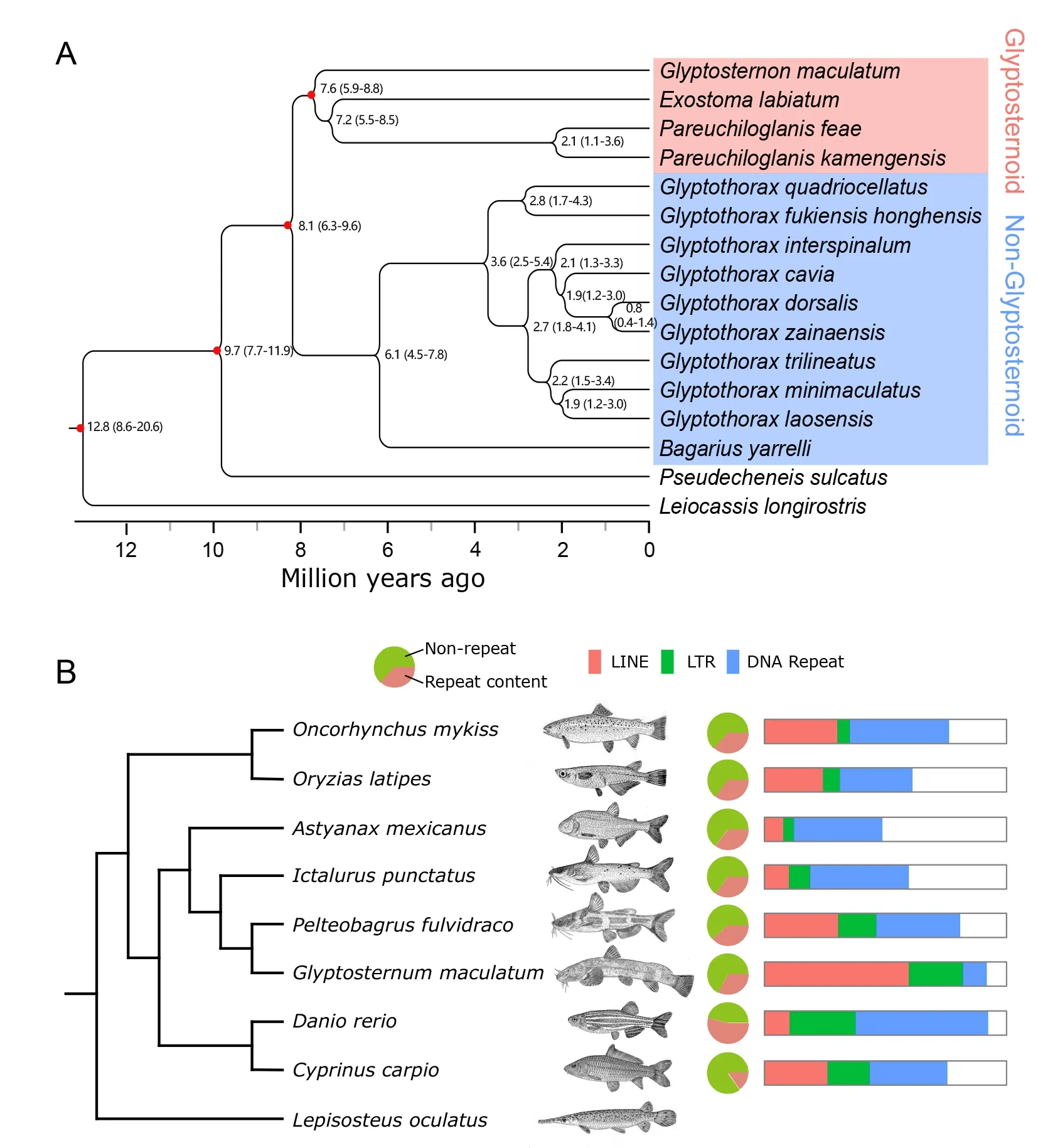
Figure 2 Phylogeny among sisorid catfish on Qinghai-Tibet Plateau and repeat content comparison of Glyptosternon maculatum to other teleosts
Expanded and selected functional genes for high-altitude environmental adaptation
Using whole-genome protein-coding gene clustering of closerelated fish species,we identified expanded gene families in theG.maculatumgenome.The functions of the expanded genes were investigated by enrichment analysis(Supplementary Tables S7,S8).We observed a large-scale expansion of gene families related to chromosome organization,including DNA packing,nucleosome organization,chromatin assembly,nucleosome assembly,DNA conformational changes,and folate metabolism(Figure 4A).
Detailed analysis revealed that genes involved in nucleosome organization were mainly core histone proteins,including H2A,H2B,H3,and H4.In theG.maculatumgenome,we identified 61,85,68,and 81 genes for the H2A,H2B,H3,and H4 histones,respectively,which were significantly higher than their copies in theP.fulvidracoandI.punetausgenomes (Figure 4B).Interestingly,most duplicated histone genes resided in the LINE/L2 elements.In contrast,few histones in theP.fulvidracoandI.punetausgenomes were found in LTR/Gypsy,simple repeat,DNA/TcMar-Tc1,and most histones did not overlap with any repetitive elements(Figure 4B).The mosaic structure of the histone with LINE/L2 demonstrated that histone proteins were expanded through the LINE/L2 bursts in theG.maculatumgenome.Strikingly,based on the timing of the expansion of the LINE/L2 elements with histone genes,we found that those repetitive elements were duplicated~2–1 Mya (Figure 4C) during the latest wave of transposon bursts,accelerated uplift of the plateau,and sudden drop in temperature (Figure 3B,C).

Figure 3 Expansion of LINE/RTE-BovB and LINE/L2 in Glyptosternon maculatum genome with tectonic movements and climate change during Qinghai-Tibet Plateau formation
In theG.maculatumgenome,many functional genes related to folate absorption and metabolism were expanded genome-wide and positively selected (Figure 4D).Two important genes,i.e.,proton-coupled folate transporter (pcft)(FDR=0.000 34) and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) transformylase/IMP cyclohydrolase(purh) (FDR=0.001 2),were positively selected in the likelihood ratio test for the nonsynonymous to synonymous substitutions (KA/KS) (Anisimova et al.,2001).Remarkably,mitochondrial monofunctional C1-tetrahydrofolate synthase(mthfd1l),dihydrofolate reductase (dhfr),adenosine kinase(adk),phosphodiesterase 3B (pde3b),and adenylate cyclase 6 (adcy6) genes,which are involved in folate metabolism and purine biosynthesis,were significantly expanded in theG.maculatumgenome (Figure 4E). We identified three mitochondrial monofunctional C1-tetrahydrofolate synthase(mthfd1l) genes with integrated functional domains in theG.maculatumgenome,but only one inP.fulvidracoandI.punetaus(Figure 4E).We showed that duplicatedmthfd1lgenes resided in the repeat rich regions,mainly the LINE RTE-BovB and L2 elements.Interestingly,those repetitive elements showed excellent synteny along the genome(Figure 4F),implying thatmthfd1lgenes may be duplicated from segmental duplications after the repeat expansion.
Population genetic diversity for G.maculatum
To investigate genetic diversity amongG. maculatumpopulations,we collected 30 samples from three sites:two from altitudes of 4 100 m (GM4100,GM2 in Figure 1) and 2 900 m a.s.l.(GM2900,GM3 in Figure 1) in the Yarlung Zangbo River and one from 4 500 m a.s.l.(GM4500,GM1 in Figure 1)in the Dogxung Zangbo River,a main branch of the Yarlung Zangbo River.Angren Lake resides in the Dogxung Zangbo River between the GM4300 and GM4500 populations (Figure 5A).
Using whole-genome resequencing data,we obtained 248 127–257 465 SNPs for species among populations(Supplementary Table S9). Sample clustering and a phylogenetic tree were constructed based on the wholegenome variations (Figure 5B,C).The GM2900,GM4100,and GM4500 populations were grouped into three clades based on PCA (Figure 5B) and phylogenetic analysis(Figure 5C).The genetic structures of the three populations further confirmed the PCA and phylogenetic analysis results(Figure 5D).UsingE.labiatumas the out-group,phylogenetic analysis based on whole-genome variants implied populations ofG.maculatummay have originated from habitats with lower altitudes in the southern QTP,consistent with previous fossil studies suggesting that glyptosternoids may have originated in southeastern Tibet and eastern Himalayan areas in Yunnan,China (Ma et al.,2015).
Using whole-genome variants among populations,we investigated the population history forG.maculatum.We found that the dynamic profiles of effective population size were similar for different populations (Figure 5E).Three populations experienced a sudden 60-fold drop in effective population size from~4 000 around 3 Mya to~60 around 2 Mya (Figure 5E).In addition,we performed a demographic analysis ofG.maculatumusing G-PhoCS (Gronau et al.,2011) to estimate the population split time and migration among populations.We showed that the GM2900 population split from their common ancestor population~10 thousand years before present (kyr BP) (Figure 5F).Remarkably,we observed that the effective population size of ancestral populations ofG.maculatumexperienced an almost 10 times reduction after the population splits,implying severe bottleneck effects during that period (Figure 5F).Furthermore,the low migration rates among populations indicated limited gene flow among populations (Figure 5F).

Figure 4 Transposon mediated expansion of histone and folate-related functional genes in Glyptosternon maculatum genome
DISCUSSION
Our phylogenetic results of sisorid catfish species using whole-genome protein-coding genes are consistent with the results obtained from mitochondrial genes,which suggest that many specialized glyptosternoid genera originated 1.9 Mya in the Pleistocene (Peng et al.,2006;Yu & He,2012),although we included moreGlyptothoraxspecies in this work.Our phylogenetic analysis also supported species radiation in the genusGlyptothorax~3 Mya.We showed thatP.feaeandP.kamengensisdiverged~2.1 Mya (Figure 2A).We also demonstrated numerous speciation events forGlyptothoraxover the last 3 Mya (Figure 2A).The times of species radiation for specialized glyptosternoid and non-glyptosternoid species are consistent with the latest sudden uplift of the QTP and Quaternary glacial period within the last 3 Mya (Jiang & Li,2014). The coincidence of massive speciation with paleogeographic and paleoclimatic events suggests that species radiation is correlated with environmental fluctuations.
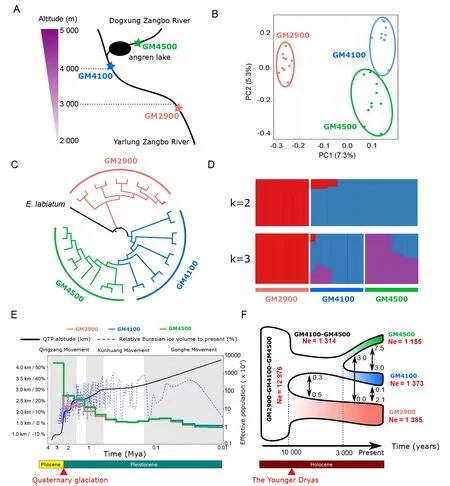
Figure 5 Population structure and history for Glyptosternon maculatum
We also observed that the content of LINEs inG.maculatumgenome,mainly RTE-BovB and L2,was significantly higher than that observed in other teleost fish(Figure 2B).Transposon elements and their associated functions have been found in teleost genomes recently.For example,Tcl-mariner transposons are thought to be involved in the rediploidization of the Atlantic salmon (Salmo salar)genome (Lien et al.,2016),and multiple bursts of LINE1,LINE2,CR1,and Deu are reported in the genomes of coelacanth (Latimeria chalumnae) (Chalopin et al.,2014) and lungfish (Neoceratodus forsteri) (Metcalfe et al.,2012).However,the distribution and roles of RTE-BovB and L2 in teleost evolution remain poorly investigated. RTE-BovB elements comprise 10.7% of the bovine genome (Adelson et al.,2009),but this type of repeat is not a dominant transposon in most teleost genomes (Supplementary Table S6).Therefore,the expansion of RTE-BovB and L2 type elements in theG.maculatumgenome is intriguing from an evolutionary perspective.Previous studies have demonstrated the potential function of transposon elements as an evolutionary driving force under rigorous environmental stress (Belyayev,2014;Craddock,2016;Platt et al.,2014).LINEs are non-LTR retrotransposons that can use self-transcribed reverse transcriptase to copy and insert their sequences with associated genes into genomes (Kazazian & Goodier,2002).Consequently,the high accumulation of LINEs in theG.maculatumgenome may be related to genome evolution and adaptation to the environment during the formation of the QTP.
Interestingly,the time estimations for the expansions of RTE-BovB and L2 type elements showed three episodic transposon bursts,illuminating another coincidence between genome evolution and time of QTP multi-stage uplift(Figure 3).Specifically,the timing of the first RTE-BovB and L2 expansion (~30 Mya) paralleled the Gangdese Movement periods in the QTP (Pan et al.,2012) (Figure 3A) and the divergence of the ancestors ofG.maculatum,I.punetaus,andP.fulvidraco(~43–37 Mya) (Supplementary Figure S2).The second RTE-BovB and L2 expansion (~20 Mya) coincided with the even more significant QTP uplift event during the Himalaya Movement at 25–15 Mya (Pan et al.,2012) (Figure 3A).The third expansion (<3 Mya) was concurrent with the sudden accelerated Tibetan uplift during the Qingzang Movement~4–1 Mya (Li et al.,2015) (Figure 3A).In addition,remarkable decreases in global surface temperature were also associated with each of the RTE-BovB and L2 bursts (Figure 3B).These drops in temperatures could significantly limit the habitat and geographically isolate endemic fish species on the QTP.Nevertheless,the episodic transposon expansions and their coincidence with multi-stage plateau uplift and climate change demonstrate the correlation between genome evolution in endemic fish and major paleogeographic and paleoclimatic events on the QTP.
We also found that the transposon expansions occurred after the ancestral divergence ofG.maculatum,I.punetaus,andP.fulvidraco(43–37 Mya;Supplementary Figure S2) and before the speciation of glyptosternoids and nonglyptosternoids (~10 Mya;Figure 3A).Thus,genomic features of earlier transposon expansions (>10 Mya) may be shared among all glyptosternoid and non-glyptosternoid species on the QTP.The most recent transposon burst occurred suddenly over a narrow window that began 4 Mya (Figure 3A).Over this last period,QTP uplift occurred at an unprecedentedly high rate,some 3–4 times that of previous events (Jiang & Li,2014).The environmental changes~3 Mya may have exerted severe adaptive selection pressure on endemic fish,as sharp transposon expansion (Figure 3A) and massive speciation(Figure 2A) were both observed during this period.
Genes related to nucleosome organization and folate metabolism were expanded in theG.maculatumgenome(Figure 4A).The main histone genes,including H2A,H2B,H3,and H4,were significantly expanded inG.maculatumcompared toP.fulvidracoandI.punetaus(Figure 4B).Histones are proteins within chromatin,and they play important structural roles in DNA packaging and structural stability (Mariño-Ramírez et al.,2005),as well as functional roles in cold stress (Verleih et al.,2015),antibiotic stimulus(Lü et al.,2014;Noga et al.,2011),UV radiation (Pawlak & Deckert,2007),and DNA repair (Schild-Poulter et al.,2003).Previous studies have demonstrated the importance of histone protein and chromatin structures to genomic stability(Oberdoerffer & Sinclair,2007).Our results indicated that LINE L2 bursts mediated the recent and rapid expansion of H2A,H2B,H3,and H4 histone genes in theG.maculatumgenome,which may be in reaction to extreme environmental adaptations to cold temperatures,food shortages,and UV exposure.Previous studies have also revealed that folate contributes to high-altitude environmental adaptation due to its important role in UV protection and DNA repair (Jablonski & Chaplin,2010) and folate-related genes exhibit significant signals of high-altitude adaptation selection in Tibetans (Yang et al.,2017).Mthfd1l,which is a mitochondrial monofunctional enzyme with 10-formyl-tetrahydrofolate (10-CHO-THF)synthase activity,plays a critical role in the folate cycle and cytoplasmic formate production (Tibbetts & Appling,2010).In human diseases,mthfd1lalso contributes to the production and accumulation of NADPH to levels that are sufficient to combat oxidative stress for cell cycle delay and apoptosis,especially in cancer cells (Lee et al.,2017).We observed the mosaic structures of histone genes andmthfd1lwithin repetitive regions,implying that LINE RTE-BovB and L2 bursts may facilitate whole-genome wide expansion of functional genes favorable for environmental adaptation on the QTP.
Glyptosternon maculatumis the only critically endangered Sisoridae species distributed at high altitudes of 2 800 m to 4 500 m a.s.l.on the QTP (Zhang et al.,2010).However,our understanding of the genetic structure and population evolution of this species on the QTP is still not clear.Using whole-genome sequencing data,we observed roughly one SNP per 2.7 kb in the genome of the wildG.maculatumpopulations,which was significantly lower than that found in theI.punetausgenome (one SNP per 93 bp) (Liu et al.,2016),thus demonstrating extremely low genetic diversity inG.maculatumpopulations.
Population analysis showed that the effective population size of ancientG.maculatumpopulations experienced a sharp 60-fold decline~2–3 Mya (Figure 5E),which coincided with the timing of the Quaternary glaciation.Quaternary glaciation,beginning 2.58 Mya,was the last major ice age to occur(Owen et al.,2008).The alternation between glaciation and interglaciation during the Quaternary glaciation period had large impact on the connectivity of the primary drainage system of rivers and lakes on the QTP (Lehmkuhl & Owen,2005),which likely lead to geographic barriers,and thus to population isolation and speciation of endemic fish on the QTP.We also found that the GM2900,GM4100,and GM4500 populations split~10 kyr BP (Figure 5F).The sudden cold and dry global climate during the Younger Dryas (11–10 kyr BP)may have contributed to the population split (Gasse et al.,1991) as the drop in temperature could have disrupted or blocked water connectivity and limited gene flow among populations during that period.Diatom records and climate studies for the Angren Lake also show an extremely low abundance of diatoms~10 kyr BP (Li et al.,1999),implying that food scarcity may also be an important reason for the population bottleneck.
CONCLUSIONS
We analyzed the genomic features and population diversity ofG.maculatum,a representative endemic fish on the QTP and the most ancient species within the subfamily Glyptosterninae.We found that whole-genome wide transposons,especially dominant repetitive elements of LINE RTE-BovB and L2,showed episodic bursts,coinciding with the timing of accelerated uplift of the QTP and dramatic climatic fluctuations. We also showed that these transposons mediated functional gene expansions,which may have contributed to the environmental adaptation ofG.maculatum.Using whole-genome variants,we determined that the ancestralG.maculatumgroup experienced two large-scale population depressions during the Quaternary glaciation~2.6 Mya and the Younger Dryas~10 kyr BP.The synchronous tectonic movements and temperature drops during glacial periods would likely have frozen habitats and disrupted drainage connectivity,leading to the historical bottleneck effects for ancientG.maculatumpopulations.For the first time,we revealed the synchronization of the genome and population evolution ofG.maculatumwith historical tectonic movements and climate events during QTP formation.Therefore,we propose that paleogeography and paleoclimate may be dominating driving forces for genome and population evolution of endemic fish on the QTP.The population wholegenome variant data provide a valuable genetic resource and opportunity to study genome and population evolution and to investigate the molecular mechanism underlying the extreme environmental adaptations of endemic fish species on the QTP.
DATA AVAILABILITY
The Hi-C sequencing data forG.maculatumwere submitted to the National Center for Biotechnology Information (NCBI)BioProject No.PRJNA447978.Genomic and transcriptome sequencing data are available from the NCBI Short Read Archive as SRR7268130–SRR7268162.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
H.P.L.designed the research.H.P.L.,S.J.X.,Z.B.M.,R.B.Y.,C.W.Z.,and S.J.X.designed and performed the experiments.S.J.X.,H.P.L.,Z.B.M.,J.Q.L.,Y.Z.,D.D.F.,S.L.Z.,M.Z.,and J.Q.L.performed repetitive element analysis.H.P.L.,Z.B.M.,and R.B.Y.contributed materials.S.J.X.,Y.Z.,and J.Q.L.created the figures.S.J.X.and H.P.L.wrote the manuscript,which was reviewed by all authors.All authors read and approved the final version of the manuscript.
杂志排行

Zoological Research的其它文章
- Melatonin relieves heat-induced spermatocyte apoptosis in mouse testes by inhibition of ATF6 and PERK signaling pathways
- 3DPhenoFish:Application for two-and threedimensional fish morphological phenotype extraction from point cloud analysis
- A new snake species of the genus Gonyosoma Wagler,1828 (Serpentes:Colubridae) from Hainan Island,China
- Inhibition of mTOR signaling by rapamycin protects photoreceptors from degeneration in rd1 mice
- A bright future for the tree shrew in neuroscience research:Summary from the inaugural Tree Shrew Users Meeting
- PINK1 gene mutation by pair truncated sgRNA/Cas9-D10A in cynomolgus monkeys