酶辅助发酵生产右旋糖酐
2021-08-15常国炜梁达奉柳颖黎志德张九花
常国炜,梁达奉,柳颖,黎志德,张九花
广东省科学院生物工程研究所,中国轻工业甘蔗制糖工程技术研究中心,广东省酶制剂与生物催化工程技术研究中心(广州 510316)
右旋糖酐是由某些微生物(如肠膜明串珠菌,Leuconostoc mesenteroides)发酵蔗糖产生的聚D-葡萄糖,其合成机理是微生物分泌右旋糖酐蔗糖酶(EC 2.4.1.5)催化蔗糖生成右旋糖酐和副产物果糖,是一条经典的蔗糖深加工路径[1]。不同分子质量的右旋糖酐有不同的应用[2],但都不乏在食品方面的应用[3-9],其中分子质量在千和万数量级的右旋糖酐(以下称为低分子质量右旋糖酐)具有益生元作用[10]。低分子质量右旋糖酐传统发酵生产的发酵效率低,目标产物比例不高,常伴有大量分子质量更高的右旋糖酐,因此发酵后需高温酸解大分子质量右旋糖酐,提高目标产物比例后再用氢氧化钠中和。“发酵-酸解-中和”三步法工艺不仅耗费大量危险化学品,还使体系引入氯离子和钠离子,增加纯化难度;酸解过程需保持高温,对设备要求高、损耗大、能耗高。针对该工艺,已有不同的改进方案,利用右旋糖酐酶(EC 3.2.1.11)酶解代替盐酸酸解[11],可形成“发酵-酶解”二步法工艺,在一定程度上优化了流程,但未能解决发酵效率低的问题;利用酶法合成低分子质量右旋糖酐[12-14],但已脱离传统发酵生产范畴,企业改造和应用成本较高。在发酵过程中添加右旋糖酐酶辅助发酵,形成“酶辅助发酵”一步法工艺,极大地优化流程,同时达到提高发酵效率和目标产物比例的目的。
1 材料与方法
1.1 材料与试剂
肠膜明串珠菌(编号GIM1.473,广东省微生物菌种保藏中心);右旋糖酐酶酶液(酶活2.053×104U/mL,广东省科学院生物工程研究所提供);右旋糖酐分子质量标准品(American Polymer Standards Corporation);蔗糖、果糖、十二水磷酸氢二钠、磷酸二氢钾、氢氧化钠、无水乙醇、无水硫酸钠、叠氮化钠(国产分析纯);细菌学蛋白胨(国产生化试剂)。
1.2 设备与仪器
LC-20A高效液相色谱仪(日本SHIMADZU);Shodex SUGAR KS-803、ShodexOHpak SB-806M HQ高效液相色谱填充柱(日本昭和电工株式会社);GRJB-5D发酵系统(镇江格瑞生物工程有限公司);等。
1.3 传统发酵和酶辅助发酵的对比
1.3.1 传统发酵操作
培养基:配制含蔗糖、细菌学蛋白胨、磷酸氢二钠、磷酸二氢钾分别为13,0.2,0.14和0.03 g/dL的溶液。
一级种子液:将40 mL培养基倒入100 mL三角瓶中,于121 ℃、20 min灭菌。接两环斜面上的菌种到三角瓶中,在摇床中培养48 h。培养条件:转速100 r/min、温度26 ℃。得到一级种子液。
二级种子液:将360 mL培养基倒入500 mL三角瓶中,按照以上步骤灭菌。将一级种子液倒入三角瓶中,在摇床中培养24 h,培养条件:转速100 r/min、温度26 ℃。得到二级种子液。
传统发酵:将3.6 L培养基倒入发酵罐中,于121℃、20 min灭菌。将二级种子液倒入发酵罐中,转速100 r/min,以0.5 mol/L NaOH溶液作为酸碱调节液,控制pH 6.5。发酵过程适时取样,按1.3.3测定蔗糖和果糖含量、醇沉物量、发酵液黏度以及右旋糖酐分子质量分布等指标。结束发酵的标志为蔗糖转化率超过90%。
1.3.2 酶辅助发酵操作
培养基、一级种子液、二级种子液同1.3.1小节。酶辅助发酵:在发酵至12 h时,加入一定量右旋糖酐酶酶液,其余操作同1.3.1小节。
1.3.3 分析方法
1.3.3.1 蔗糖、果糖的测定
将蔗糖和果糖置于96 ℃烘箱中干燥2 h[15],用超纯水配制质量浓度分别为0.6,1.2,1.8,2.4和3.0 g/dL的蔗糖和果糖溶液,用液相色谱进行测定,色谱条件:以经0.45 μm膜过滤的超纯水为流动相;Shodex SUGAR KS-803色谱柱;柱温50 ℃;流速1.0 mL/min;进样量10 μL;检测器为示差折光检测器。以峰高对浓度制得标准曲线的线性回归方程(过原点)。蔗糖标曲为h=70191c,R2>0.9999;果糖标曲为h=61006c,R2>0.9999。
将发酵液稀释适当倍数(V/V),经0.45 μm膜过滤后按上述条件进行液相色谱分析,通过标准曲线和稀释倍数计算出蔗糖、果糖含量。
1.3.3.2 醇沉物质量的测定
取3 mL发酵液,加入6 mL无水乙醇,剧烈振荡后,以8000 r/min离心5 min,弃上清液,用滤纸轻轻吸干管内壁和沉淀表面液体,得出醇沉物的质量。
1.3.3.3 黏度的测定
加入发酵液至10 mL ep管的管口,倒置ep管使液体流出,待液体流出至10 s无液滴滴出,测得残留在管内的液体质量,用该质量代表发酵液黏稠程度。设定:小于0.08 g,为低黏度;0.08~0.24 g,为中黏度;大于0.24 g,为高黏度。
1.3.3.4 右旋糖酐分子质量分布的测定
测定方法为高效液相色谱法,具体步骤和条件参照文献[16]。所用色谱柱为Shodex OHpak SB-806M HQ色谱柱,理论板数计算公式见式1(源于色谱柱使用说明书)。经试验,得出理论板数为N=5.54×(21.300÷0.494)2=10299>5000,说明柱子效能正常,可用于右旋糖酐分子质量测定。分子质量标准品选用720000,95000,53000和43000 g/mol,得到分子质量标准曲线lgMW=-0.6857tR+16.80。用两倍浓度的流动相稀释发酵液2倍后进行测定。

式中:N为理论板数;tR为出峰时间,min;W为半峰宽度,min。
1.4 酶辅助发酵不同加酶方案对产物的影响
酶辅助发酵操作按1.3.2小节进行,其中加酶方案按表1进行;右旋糖酐分子质量分布的测定按1.3.3.4小节进行。
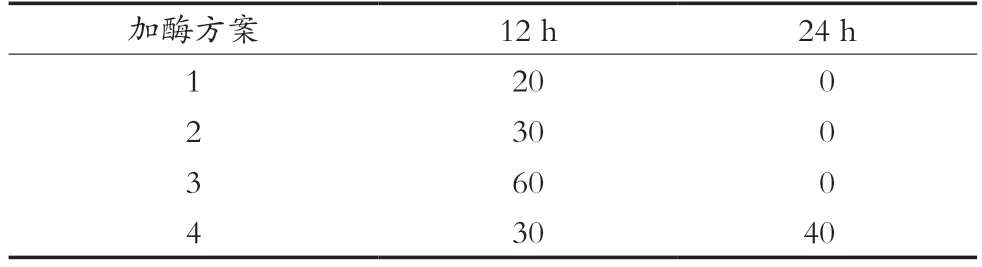
表1 酶辅助发酵不同加酶方案 单位:μL
2 结果与讨论
2.1 传统发酵和酶辅助发酵的对比分析
由图1~图3可知,两种发酵工艺前12 h的情况是相同的。前10 h蔗糖含量变化不大,蔗糖转化率处于较低水平(约5%),果糖和醇沉物量也没有明显变化,说明各主要成分均无明显变化,是典型的微生物生长迟缓期。到了12 h,蔗糖转化率提高至11.9%,说明在10~12 h,碳源利用速度加快。此时果糖和醇沉物量仍无明显变化,可能是因为碳源主要用于菌体生长繁殖,用于合成右旋糖酐的右旋糖酐蔗糖酶未大量分泌,所以右旋糖酐未被大量合成,副产物果糖未大量生成。前12 h的发酵液黏度处于低黏度阶段(如表2所示),因为菌体和右旋糖酐的含量均较低,此时黏度主要来源应是蔗糖。
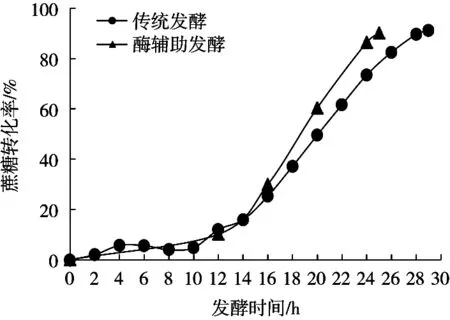
图1 两种工艺蔗糖转化率对比
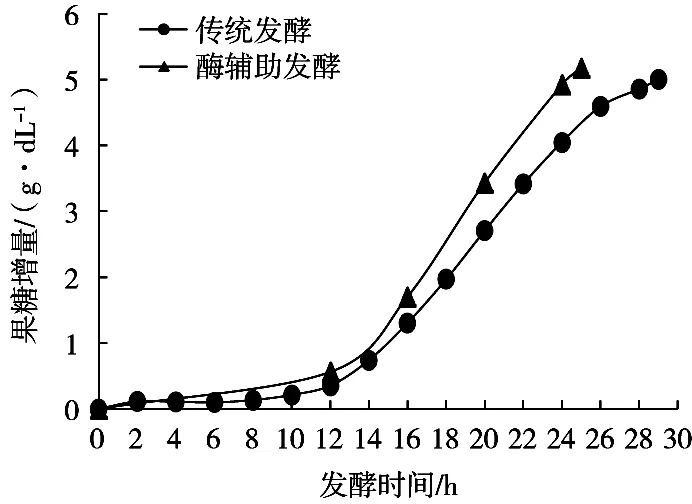
图2 两种工艺果糖增量
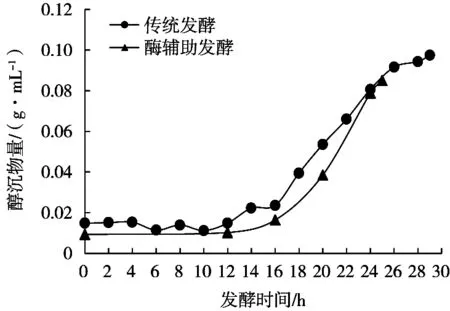
图3 两种工艺醇沉物质量对比
两种发酵工艺主要区别在发酵12 h后。通过图2和图3可知,传统发酵至14 h,果糖量和醇沉物量有明显上升,说明右旋糖酐开始被大量合成。从蔗糖加速消耗至果糖、醇沉物量明显上升有约2 h的时间差,这可认为是微生物从生长迟缓期慢慢进入酶系活跃、代谢旺盛的对数生长期的转变特征。与此同时,发酵液黏度增加到中等黏度。
发酵至20 h,蔗糖转化率达50%,醇沉物量和果糖量不断增大,意味着右旋糖酐含量不断增大。在取样过程中可明显感到流动性变差,进行实验操作时,难以使用如移液管、移液枪等工具准确量取体积,发酵液黏度达到了高黏度阶段。到了28 h,蔗糖转化率达89.6%,从图1可看出蔗糖转化开始减慢;到29 h,蔗糖转化率达91.2%,结束发酵,估算出蔗糖转化率达90%的时间约28.3 h。根据果糖增量可以计算出结束发酵时右旋糖酐产量(数据见表2),此时产物与底物比(发酵结束时的右旋糖酐生成量:蔗糖消耗量)为37.83%(理想最大值为47.37%,即蔗糖全部用于合成右旋糖酐)。
上述过程是传统发酵的情况,而酶辅助发酵则是在12 h添加右旋糖酐酶,其直接作用的是水解发酵体系中的右旋糖酐的α-1,6糖苷键,降低其分子质量。由图1和图2可知,在12 h后,酶辅助发酵蔗糖转化率和果糖增速明显高于传统发酵,达90%的时间仅用25.0 h,比传统发酵缩短3.3 h,即发酵周期缩短11.7%。通过表2可知,酶辅助发酵的低黏度期延长了6 h,并将中黏度期延长至发酵结束;在12~24 h,酶辅助发酵的蔗糖消耗速率和果糖增长速率分别提高了21.4%和18.2%;右旋糖酐产量提高了3.5%;产物与底物比增加2.64个百分点,至40.47%。综上分析,酶辅助发酵由于右旋糖酐酶作用,使右旋糖酐分子质量下降,发酵体系黏度降低,改善了发酵环境,提高了底物利用效率和产物生成效率。

表2 两种发酵工艺数据对比
从图3可知,发酵25 h时,两个工艺醇沉物量相差不大,是由于1.3.3.2小节所用的乙醇量不大,比例为1∶2(发酵液∶无水乙醇),且沉淀时间较短,这均不利于低分子质量右旋糖酐沉淀析出。所以看似相近的数据,实际上酶辅助发酵的右旋糖酐量应该更多。
2.2 传统发酵和酶辅助发酵产物分子质量分布对比
表3详细地列出了不同发酵工艺和加酶方案发酵结束时产物分子质量分布情况,不同数量级的比例为高效液相色谱图中对应保留时间区间积分面积的比例,表3中所有数据均没有将百级的产物纳入统计和计算。传统发酵所得右旋糖酐分布极分散,最高达千万级。低分子质量右旋糖酐比例只有54.2%,还有45.8%的大分子质量右旋糖酐,这部分右旋糖酐发酵液黏度增大,影响发酵效率,且不仅无法直接利用,还需要通过“酸解-中和”工序或“酶解”工序进行水解来提高目标产物比例,所需水解条件较强[17]。
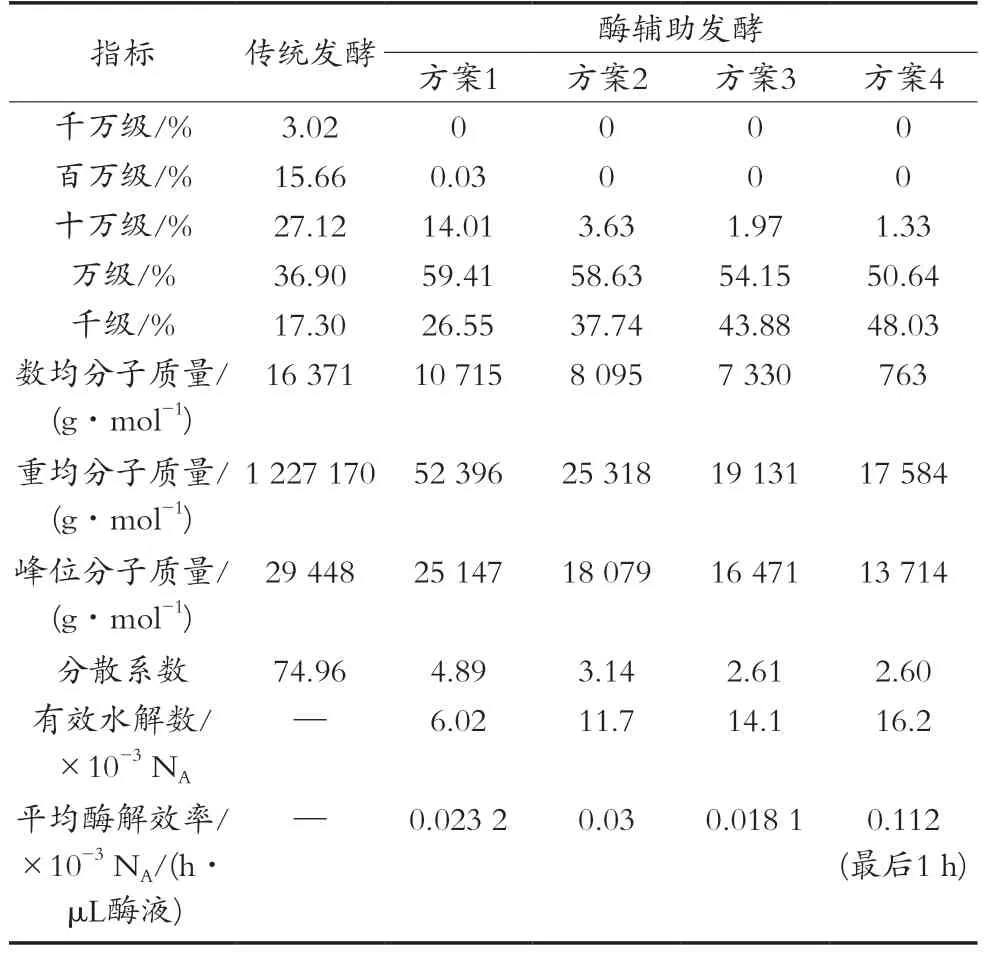
表3 各工艺产物分子质量分布
酶辅助发酵中,右旋糖酐酶添加量仅为每升发酵液几至十几微升酶液,发酵结束时目标产物比例上升至86%~99%,仅含比例不高的十万级右旋糖酐,分散系数从74.96下降至5以下,效果极显著。根据式(2)可以计算出右旋糖酐酶的有效水解数,不同方案的有效水解效率有所差异,均值为0.0238×10-3NA/(h·μL酶液),该数据可指导酶辅助发酵加酶量控制。
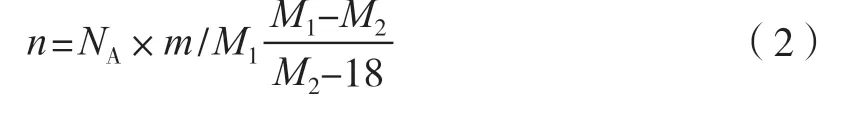
式中:n为有效水解数;NA为阿伏伽德罗常量;m为右旋糖酐质量,g;M1为水解前右旋糖酐数均分子质量,g/mol;M2为水解后右旋糖酐数均分子质量,g/mol;18为水的摩尔质量,g/mol。
根据方案2和方案4的数据可知,在一次加酶辅助发酵基础上,在24 h再次加入酶液,可形成二次加酶辅助发酵工艺。通过有效水解数可计算出,二次加酶辅助发酵工艺最后1 h平均酶解效率达0.112×10-3NA/(h·μL酶液),是一次加酶辅助发酵的4.7倍,说明该工艺可在结束发酵前1 h进一步调节右旋糖酐分子质量分布,为生产带来一定的灵活性。综上,酶辅助发酵仅需极少量右旋糖酐酶液,就能显著提高目标产物比例,大幅降低分散系数,使产物分子质量更加集中,并可通过多次加酶灵活调节产物分子质量分布。
3 结论
在发酵生产低分子质量右旋糖酐过程中,添加极少量右旋糖酐酶,可形成“酶辅助发酵”工艺。酶辅助发酵中,右旋糖酐酶能高效水解大分子质量右旋糖酐,降低发酵液黏度,改善发酵环境,从而提高发酵效率,使蔗糖转化率达90%的发酵周期比传统发酵缩短11.7%,右旋糖酐产量提高3.5%,目标产物比例提高至90%以上,且分布集中。多次加酶辅助发酵还可灵活控制产物分子质量分布。酶辅助发酵省去传统发酵后续的酸解和中和工段,极大地优化流程,达到节能减排的目的。
