苍白球黑质红核色素变性1例及文献复习*
2021-08-13南在元冯德琳刘皓珏于佳辉王琳琳
南在元,冯德琳,刘皓珏,于佳辉,王琳琳
1.黑龙江省哈尔滨市儿童医院神经内科,黑龙江哈尔滨 150010;2.黑龙江省哈尔滨市儿童医院中医针灸科,黑龙江哈尔滨 150010;3.黑龙江省哈尔滨市妇幼保健院麻醉科,黑龙江哈尔滨 150010
苍白球黑质红核色素变性为脑组织铁沉积性神经变性病(NBIA),由基因突变导致椎体外系症状为主,伴其他复杂临床症状,在脑组织特定部位可见异常铁沉积[1]。在儿科临床工作中对于颅脑影像学检查的特点很大程度上依赖于医学影像科的报告,而影像报告中未提示具有的特征性术语或可能的疾病时容易导致漏诊或误诊,对于罕见病更是如此,现分享1例苍白球黑质红核色素变性,总结其临床及影像学特点。
1 病例资料
患儿,女,4岁11月龄,主因“发作性头颈向右扭转1周”入院,患儿运动发育落后(23月会走,走路不稳,易跌倒)、语言发育落后(至今发音不准),家长否认存在家族史。患儿在住院前1周开始出现不自主头颈向右侧扭转,经家长言语或触碰干扰可停止,每日发作数次逐渐增多、发作时间延长,严重时可因头颈扭转导致原地转圈,直至摔倒。
查体:生命体征平稳,周身无皮疹,心、肺、腹无异常,意识清楚,精神正常,构音不清,四肢肌张力增高,四肢肌力V级,双侧膝腱反射及跟腱反射亢进,双侧巴氏征阳性,脑膜刺激征阴性,共济试验笨拙。颅脑MRI检查:双侧基底节对称性稍长T1稍长T2信号(图1)。脊髓MRI检查:无异常。血常规、尿常规、心肌酶、肝功能、肾功能、血离子、血氨、血乳酸、甲状腺功能、甲状旁腺激素检测:均正常。裂隙灯下未见K-F环。眼科眼底检查:视网膜色素紊乱。基因检测:患儿系泛酸激酶2(PANK2)基因c.796-797del和c.1555T>C突变的复杂杂合子(c.796-797del:父亲未发现变异,母亲杂合变异;c.1555T>C:父亲杂合,目前未发现变异)。致病性报道:c.796-797del,为致病变异(图2);c.1555T>C,为可疑致病变异(图3)。

注:T2图像呈现“虎眼征”,即双侧苍白球对称性中央内侧高信号,周边低信号。
治疗:口服左旋多巴(第1周,半片/次,每日2次;第2周,1片/次,每日2次),第7天症状明显改善(无发作性头颈扭转症状),3个月及6个月后随访,无明显临床症状。
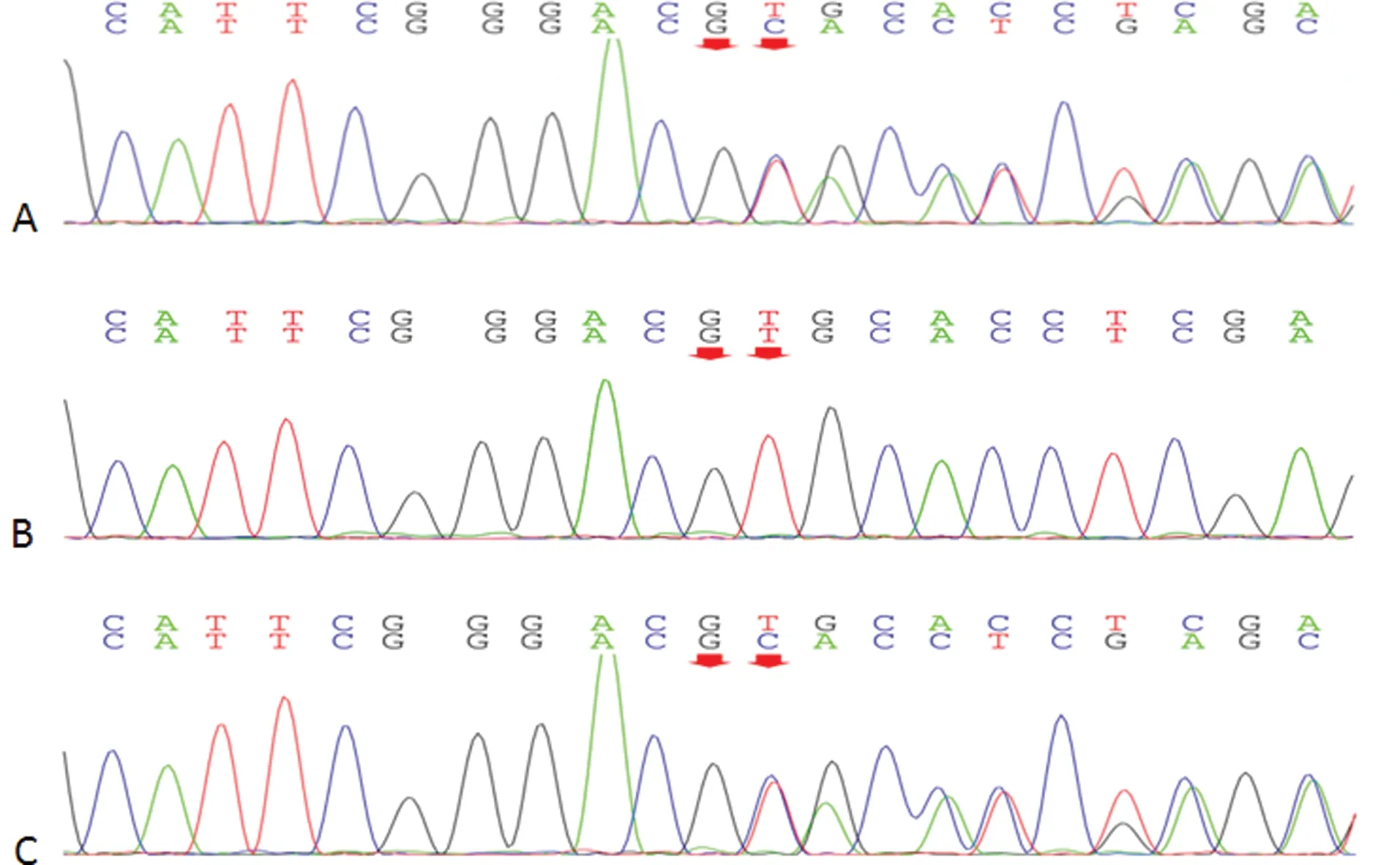
注:A显示患儿PANK2基因(chr20:3888740)c.796-797del;B显示其父该位点正常;C显示其母该位点杂合变异。

注:A显示患儿PANK2基因(chr20:3899336)c.1555T>C;B显示其父该位点杂合变异;C显示其母该位点正常。
2 讨 论
NBIA疾病谱中10个亚型已明确致病基因,各亚型采用“突变蛋白相关性神经变性病”模式统一命名[2]。苍白球黑质红核色素变性是在1922年由HALLERVORDEN和SPATZ首次报道[3],故又称Hallervorden-Spatz病(HSD),其致病基因定位于20P12.3-P13,包含7个外显子,相对分子质量为1.85×103,编码蛋白为PANK2蛋白,故命名为泛酸激酶相关性神经变性病。
研究显示,PANK2蛋白存在于线粒体[4]。动物实验显示,PANK2基因突变的纤维母细胞及PANK2基因敲除小鼠和果蝇模型存在线粒体功能缺陷,包括线粒体膜电位降低、线粒体肿胀和线粒体嵴改变等[5]。PANK2基因参与辅酶A的生物合成,其与辅酶A和酰基辅酶A之间存在负性调控。PANK2基因突变使泛酸磷酸化不能进行,引起胱氨酸蓄积,螯合铁并沉积,病理表现为苍白球和黑质网状区及一些邻近区域存在大量色素聚集,主要为含铁色素;神经轴索呈球形体肿胀,神经元脱失,铁沉积和胶质细胞增生[6]。PANK2基因突变还可引起线粒体特异性脂肪酸合成途径破坏,有研究报道,PANK2基因突变与脂肪合成缺陷相关[7]。也有研究报道,部分患者包括来自常染色体隐性遗传家系的患者,没有发现PANK2基因突变,表明HSD具有很强的遗传异质性[8]。
HSD的临床表现多样,主要表现为锥体外系症状,如肌张力障碍、震颤、舞蹈手足徐动症、肌强直、运动迟缓等,随病情进展可逐渐出现痉挛步态、腱反射增高、病理征阳性等锥体束受累症状,同时可伴有构音不清、吞咽困难、性格改变、视神经萎缩及视网膜色素变性等,亦有部分患者始终仅表现单一的症状[9]。本病虽以儿童和20岁前青少年多见,但有相关文献报道最大发病年龄为80岁[10]。颅脑MRI检查对HSD的诊断有重要价值[11],在T2WI影像中双侧苍白球部位由于铁沉积显示低信号,而苍白球的前内侧由于神经元死亡、胶质增生而显示高信号,称“虎眼征”。晚期患者由于铁沉积进一步加重,中间高信号逐渐消失,呈较均一低信号。临床分两种类型:(1)典型HSD,10岁前发病,多在15年内不能行走,20岁前生活不能自理,多于35岁前死亡;(2)非典型HSD,10岁后发病,病程进展较前者缓慢,智力减退不显著,无家族史。典型HSD均有PANK2基因突变,MRI表现为“虎眼征”。不典型HSD中约1/3患者有PANK2基因突变,MRI表现为“虎眼征”;而没有PANK2基因突变的不典型患者MRI没有“虎眼征”[1]。
目前对NBIA缺乏有效的治疗手段,治疗包括药物治疗、手术治疗及铁离子螯合剂等,均为对症治疗,表现为肌张力增高和运动迟缓,可用左旋多巴缓解症状;痫性发作可用抗癫痫药物;用抗抑郁药改善患者情绪等[1]。
本病例为PANK2基因突变的复杂杂合子,其中c.796-797del为既往文献未曾报道过的基因突变类型,提示第266个氨基酸开始其合成发生改变,并在改变后的第25个氨基酸终止;其中c.1555T>C已有文献报道,提示第519个氨基酸由苯丙氨酸变为亮氨酸,两个突变分别来自父亲和母亲,但父母无临床症状,说明两个致病突变的共同作用引起蛋白功能异常。根据发病年龄、临床症状及检查结果,均符合典型HSD,但临床中值得注意的是,影像报告中未提及术语“虎眼征”时,临床医师诊断HSD仍较困难,这要求临床医师有较高的阅片能力及文献查阅能力。本病例给予左旋多巴对症治疗后临床症状改善显著,但仍需持续随访观察。铁离子螯合剂对本病的治疗有些争议,但近些年有报道指出使用铁离子螯合剂治疗HSD取得了较满意的疗效[12],故将来可能成为治疗HSD的一种有效手段。
