Integration of association and computational methods reveals functional variants of LEPR gene for abdominal fat content in chickens
2021-08-12LIYudongWANGWeijiaLIZiweiWANGNingXIAOFanGAOHaiheGUOHuaishunLIHuiWANGShouzhi
LI Yu-dong ,WANG Wei-jia ,LI Zi-wei,WANG Ning,XIAO Fan,GAO Hai-he,GUO Huaishun,LI Hui,WANG Shou-zhi
1 Key Laboratory of Chicken Genetics and Breeding,Ministry of Agriculture and Rural Affairs,Harbin 150030,P.R.China
2 Key Laboratory of Animal Genetics,Breeding and Reproduction,Education Department of Heilongjiang Province,Harbin 150030,P.R.China
3 School of Animal Sciences and Technology,Northeast Agricultural University,Harbin 150030,P.R.China
4 Fujian Sunnzer Biotechnology Development Co.,Ltd.,Guangze 354100,P.R.China
Abstract Leptin receptor (LEPR) plays a vital role in obesity in humans and animals.The objective of this study is to assess LEPR functional variants for chicken adipose deposition by integration of association and in-silico analysis using a unique chicken population,the Northeast Agricultural University broiler lines divergently selected for abdominal fat content (NEAUHLF).Five online bioinformatics tools were used to predict the functionality of the single nucleotide polymorphisms (SNPs) in coding region.Further,the possible structure–function relationship of high confidence SNPs was determined by bioinformatics analyses,including the conservation and stability analysis based on amino acid residues,prediction of protein ligand-binding sites,and the superposition of protein tertiary structure.Meanwhile,we analyzed the association between abdominal fat traits and 20 polymorphisms of chicken LEPR gene.The integrated results showed that rs731962924 (N867I) and rs13684622(C1002R) could lead to striking changes in the structure and function of proteins,of which rs13684622 (C1002R) was significantly associated with abdominal fat weight (AFW,P=0.0413) and abdominal fat percentage (AFP,P=0.0260) in chickens.Therefore,we are of the opinion that rs13684622 (C1002R) may be an essential functional SNP affecting chicken abdominal fat deposition,and potentially applied to improvement of broiler abdominal fat in molecular marker-assisted selection (MAS) program.Additionally,the coupling of association with computer electronic predictive analysis provides a new avenue to identify important molecular markers for breeders.
Keywords:chicken,LEPR,nsSNPs,bioinformatics tools,abdominal fat content,association analysis
1.Introduction
Poultry breeding,especially broiler breeding,has made remarkable achievements in the past half a century.The daily gain,feed conversion rate,and disease resistance of broilers have been significantly improved.However,accompanied with intensive selection of the growth rate of broilers,the excessive deposition of body fat in broilers,especially abdominal fat,has also become a prominent problem in the poultry industry (Wanget al.2004).For broilers,excessive body fat leads to a reduced feed conversion rate and low economic value (Tianet al.2010;Donget al.2015).Therefore,it’s an urgent problem to be solved in the broiler industry to control the excessive accumulation of abdominal fat,and further improve the feed conversion rate and meat quality of broilers.Chicken abdominal fat,however,is a complex trait controlled by multiple genetic and environmental factors,and its measurement is costly and laborious by slaughtering birds,which largely hinders genetic improvement based upon birds’ abdominal fat measures.Marker-assisted selection(MAS) is one of the most effective methods to tackle this issue.
The identification of functional single nucleotide polymorphisms (SNPs) for complex disease or important economic traits is one of the research hotspots in humans and animals.As far as research strategies are concerned,two main strategies,such as experimental andin-silicostrategies are widely applied to identify functional variants for complex traits.Over the past few years,usinginsilicotools to predict damaging or functional SNPs has been an efficient approach requiring less time and cost than experimental procedures,and preliminary screened damaging or functional SNPs are candidates for subsequent functional verification experiments (Zhang Met al.2020).
Some SNPs within gene coding region can lead to changes in the peptide sequence,which are called nonsynonymous SNPs (nsSNPs).nsSNPs are important factors leading to the functional diversity of candidate genes in animal populations.The functional prediction of nsSNPs based on bioinformatic tools will help us to better understand the relationship between observed phenotypic variation and genotype (Falomir-Lockhartet al.2018).More recently,researchers have carried out fruitful work on the analysis and prediction of functional gene mutations affecting animal traits of interest,such as screening for nsSNPs associated with bovine mastitis resistance (Jacobet al.2020) and identifying the nsSNPs ofKITgene associated with grey phenotype in alpacas (Joneset al.2019)viacomputational tools.Nevertheless,considering the fact that SNPs used in this kind of research were derived from public databases of human and animal,functional SNPs identified would not always effective in other populations due to different genetic backgrounds,which could impede their application.An efficient way to overcome this shortcoming would be thatinsilicoanalysis and association of SNP with traits or diseases of interest are integrated to characterize functionality of SNPs in target gene.
Leptin receptor (LEPR) is a high-affinity receptor of leptin and has a single transmembrane structure.Leptin binds its receptorLEPRdirectly,to transfer the signal into muscle cells,and activate the fat metabolism pathway in muscle cells,and lead to the enhancement of fatty acid oxidation metabolism in muscle.Many studies in mice and humans revealed that there is a close relationship between theLEPRgene and abdominal fat content.Allensworth-Jameset al.(2015) found that ablation ofLEPRcaused severe growth hormone deficiency and abdominal obesity in male mice.Foucanet al.(2019) suggested an influence of K656N polymorphism in theLEPRgene on abdominal obesity in this Afro-Caribbean population of Guadeloupe Island.Similarly,chickenLEPRplays a vital role in this signal transduction pathway,which is related to the deposition and distribution of fat (El Moujahidet al.2014;Leiet al.2015).Leiet al.(2015) demonstrated that active immunization against chicken leptin receptor stimulates metabolism and reduces abdominal fat deposition in growing chickens.In recent studies,expression of leptin andLEPRmRNA have been discovered in chicken duodenum,suggesting thatLEPRof chicken also has a regulatory effect on appetite in the short term (Seroussiet al.2019).Previous studies have shown that some SNPs ofLEPRgene were associated with production traits in chickens.For example,nucleotide mutations in intron 8 and exon 9 of chickenLEPRgene were significantly associated with phenotypic variations in birth weight,abdominal fat weight (AFW),abdominal fat percentage (AFP),and liver weight (Guet al.2002;Wang S Zet al.2019).
Up to now,however,there is a lack of comprehensive investigation of functionality of SNPs withinLEPRgene,especially in coding region,where the functional consequences of the changed amino acid caused by SNPs remain largely unclear.It was hypothesized that there would be some functional SNPs,in exons of chickenLEPRgene in relation to AFW and AFP.The purpose of this study is to identify the functional SNPs within the exon region ofLEPRgene related to chicken fat deposition using bothin-silicoapproach and association analysis.
2.Materials and methods
2.1.Experimental populations and phenotypic measurements
The Northeast Agricultural University broiler lines divergently selected for abdominal fat content (NEAUHLF) have been selected since 1996 using percentage of abdominal fat and plasma very low-density lipoprotein concentration as selection criteria.The G0generation of the 2 lines came from the same grandsire line originating from the Arbor Acres broiler,which was then divided into 2 lines according to plasma very low-density lipoprotein concentration at 7 weeks of age (Lenget al.2009).The experimental materials of this study were derived from the 19th generation (G19)broilers of NEAUHLF.A total of 329 cocks (159 individuals of fat line and 170 individuals of lean line) were used in this study.These birds were kept under the same environmental conditions.The temperature in the henhouse was kept at 18 to 25°C,and the air humidity was maintained at 60 to 65%.The birds of each line were raised in two hatches and housed in pens.All birds had free access to feed and waterad libitum.Commercial corn-soybean-based diets that met all nutrient requirements of broilers recommended by National Research Council (NRC 1994) were provided to the birds.From hatch to 3 weeks of age,the birds received a starter feed (metabolizable energy (ME),3 000 kcal kg–1;crude protein (CP),210 g kg–1) and from 4 weeks of age to slaughter the birds were fed a grower diet (ME,3 100 kcal kg–1;CP,190 g kg–1).The live weight (body weight at 7 weeks of age,BW7) was measured before slaughtering at 7 weeks of age.All birds were slaughtered by cervical dislocation and exsanguination from the jugular vein.Then abdominal fat weight (AFW,g) was manually separated and weighed and abdominal fat percentage (AFP (%)=(AFW(g)/Body weight (g))×100) was calculated according to the performance terms and measurement for poultry formulated by Chenet al.(2004) .
2.2.SNP data
We constructed individual 350 bp DNA libraries for 329 cocks of G19 of NEAUHLF,and carried out whole-genome re-sequencing.A total amount of 1.5 μg DNA of each sample was taken as the input material for sample preparation.The Truseq Nano DNA HT Sample Preparation Kit (Illumina,USA) was used to generate a sequencing library and index codes were added to the attribute sequence of each sample.After acoustic degradation,DNA fragments were endpolished,A-tailed,and ligated with the full-length adaptor for Illumina sequencing with further PCR amplification.Then the PCR products were purified with AMPure XP System (Beckman Coulter,Beverly,CA,USA).The size distribution of the library was analyzed by Agilent2100 Biological Analyzer (Agilent,Santa Clara,CA,USA) and quantified by real-time PCR (Applied Biosystems,USA)(Zhang Het al.2020).
After alignment,variant calling was performed for all samples by using the Unified Genotyper function in GATK 3.3 Software.SNPs were selected by using the VariantFiltration parameter in GATK.Detailed description is presented in report by Zhang Het al.(2020).A total of 20 SNPs onLEPRexons (average coverage of 5.3-fold),were obtained,and their genotypes in G19 are available from re-sequencing.
2.3.Screening functional nsSNPs with five in-silico analysis tools
Sorts intolerant from tolerant (SIFT) (http://sift.jcvi.org/) is a sequence homology-based tool that predicts variation in protein function caused by the change in amino acid sequence.Computationalin-silicoanalysis using SIFT can predict 90% of damaging SNPs.The functional consequences of amino acid substitutions caused due to nsSNPs were ascertained using the respective SIFT score.
PhD-SNP is a Support Vector Machine (SVM) which uses evolutionary information to sort out SNPs related to Mendelian and complex diseases from the neutral ones(Elkhattabiet al.2019)
SNPs&GO is also a SVM method that can accurately predict disease-related mutations from protein sequences(Portoet al.2015).The input is the FASTA sequence of the whole protein,while the output is based on the differences between the neutral and disease-related variations of the protein sequence (Capriottiet al.2013).
PolyPhen-2.0 (http://genetics.bwh.harvard.edu/pph2/uses) is an iterative algorithm that uses straightforward comparative and physical considerations to predict the possible impact of the substitution of an amino acid on the function and structure of a protein.
SNAP,which predicts the function of mutations,is based on a machine-learning device called a neural network.It determines the effect of non-synonymous SNPs by considering various sequence and variation characteristics.
2.4.Effect of amino acid substitutions on mutant protein stability
We used I Mutant 3.0 and MUpro servers to study the effects of amino acid substitution on the stability of mutant proteins.Stability change was expressed as DDG value in kcal mol–1at 25°C and pH 7.The server performs a structure-based analysis of mutant proteins that are replaced on a single amino acid residue and provides estimates of free energy changes in mutant proteins (Alshatwiet al.2012;Arshadet al.2018;Khanet al.2018).
2.5.Analysis of conserved residues in chicken LEPR protein
The Protein BLAST on NCBI online website was used to search for highly-homologous amino acid sequences ofLEPRin different species,then Clustal-Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) and Jalview Software were used for multiple sequences alignment.ConSurf (http://consurftest.tau.ac.il) was used to associate SNPs with highly conserved buried and exposed amino acid residues in LEPR protein (Barr 2014;Nailwal and Chauhan 2017;Badgujaret al.2019).The common point of these two methods is to look for conserved amino acid sequence sites,the more conservative locus mutation,the more likely theLEPRfunction or structure on the impact of the protein.
2.6.Prediction of disease-related amino acid substitution and phenotypes by MutPred2
MutPred2 is a standalone and web application developed to classify amino acid substitutions as pathogenic or benign,predicting the pathogenicity of amino acid substitutions and their molecular mechanisms.It also predicts their impact on over 50 different protein properties,so that the molecular mechanism of pathogenicity can be inferred (Amiret al.2018;Arshadet al.2018).
2.7.Prediction of ligand binding site with FTSite server
FTSite (http://ftsite.bu.edu) is an online web server that predicts the ligand-binding sites of proteins with high accuracy (Singh and Mahalingam 2017).FTSite server was performed to find whether or not the identified nsSNPs present in LEPR protein-binding region (Salehet al.2016).
2.8.Comparison of secondary structures and homology modeling for structure prediction
SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html) is a second-order protein prediction method based on the existing database,while SOPMA is its improved version,which can be self-optimized based on increasing the database to improve the prediction accuracy of the second-order protein structure (Agrahariet al.2019;Islamet al.2019;Wang Qet al.2019).
MODELLER v9.23 is a program for comparative protein structure modeling by satisfaction of spatial restraints and can calculate a model containing all non-hydrogen atoms automatically.The user provides an alignment of a sequence with known related structures for homology or comparative modeling of three-dimensional protein structures (Guzziet al.2020).The generated structural model was selected and subjected for the structural validation using PROCHECK v3.5 online tool (the quality of the protein structure was correctly evaluated by analyzing residue-by-residue geometry and whole structure geometry) (AbdulAzeezet al.2016;Momenet al.2017).Using the ModRefiner tool of the I-TASSER online website for structural optimization and energy minimization (Xu and Zhang 2011;Dakalet al.2017;Guttulaet al.2019).The amino acid residue substitutions or mutant structures were generated using the VMD Software.GLSL coloring method was used to highlight the three-dimensional structure of the sequence near the mutant amino acid site(Gomeset al.2012).The TM-align online tool (https://zhanglab.ccmb.med.umich.edu/TM-align/) is used for protein structure alignment and RMSD value calculation (Rasalet al.2015;Dakalet al.2017).
2.9.Association analysis of LEPR gene polymorphisms
JMP Software was used to analyze the association between polymorphic loci and abdominal fat traits.The following mixed linear model was established according to data types and population characteristics:

where Y is the observed value of the character (AFW and AFP),μ is the average value of the population,Sire (Line) is the random effect of Sire nested within the Line,Dam (Line,Sire) is the random effect of Dam nested within Sire and Line,G is the fixed effect of the SNP genotypes,Line is the fixed effect of the Line,BW7is the covariate for AFW,and e is the residual random error.Each individual was the experimental unit.P<0.05 was considered statistically significant.Multiple comparisons between least squares means of the different genotypes were performed by Tukey HSD.
3.Results
3.1.Distribution of SNP on LEPR gene
Sequencing results revealed that a total of 20 nsSNPs were on the exon ofLEPRgene.After these SNPs were matched to the dbSNP Database,we found that these 20 nsSNPs were known loci,then they were named with known ID.The genomic structure of the entireLEPRgene contains 20 exons,spanning 30.0 kb (Fig.1).
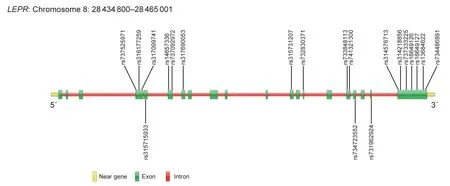
Fig.1 Distribution diagram of LEPR gene non-synonymous SNPs (nsSNPs) in chickens.
3.2.Screening of nsSNPs based on functional analysis
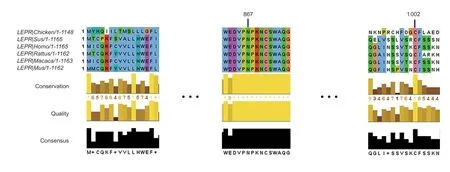
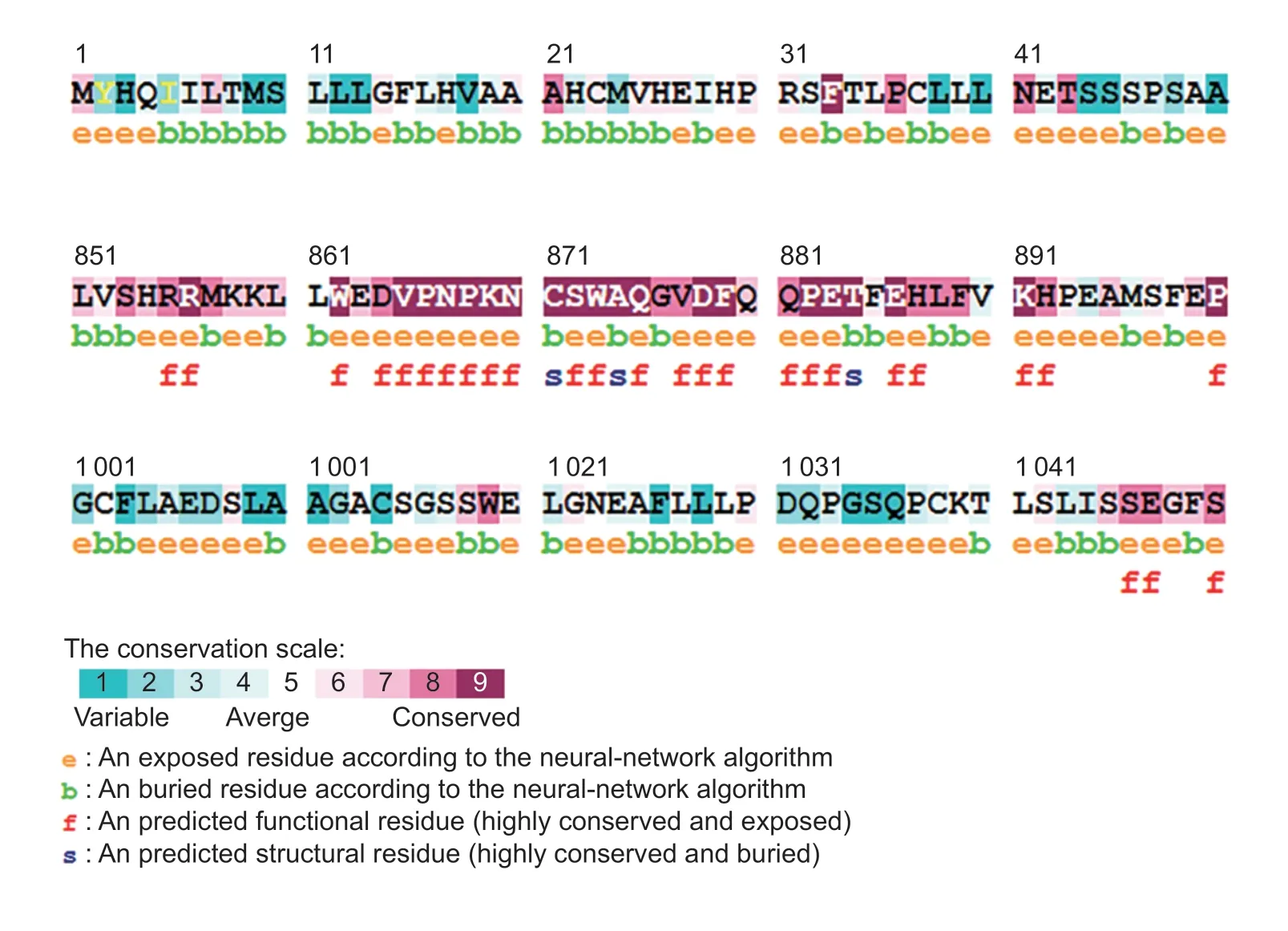
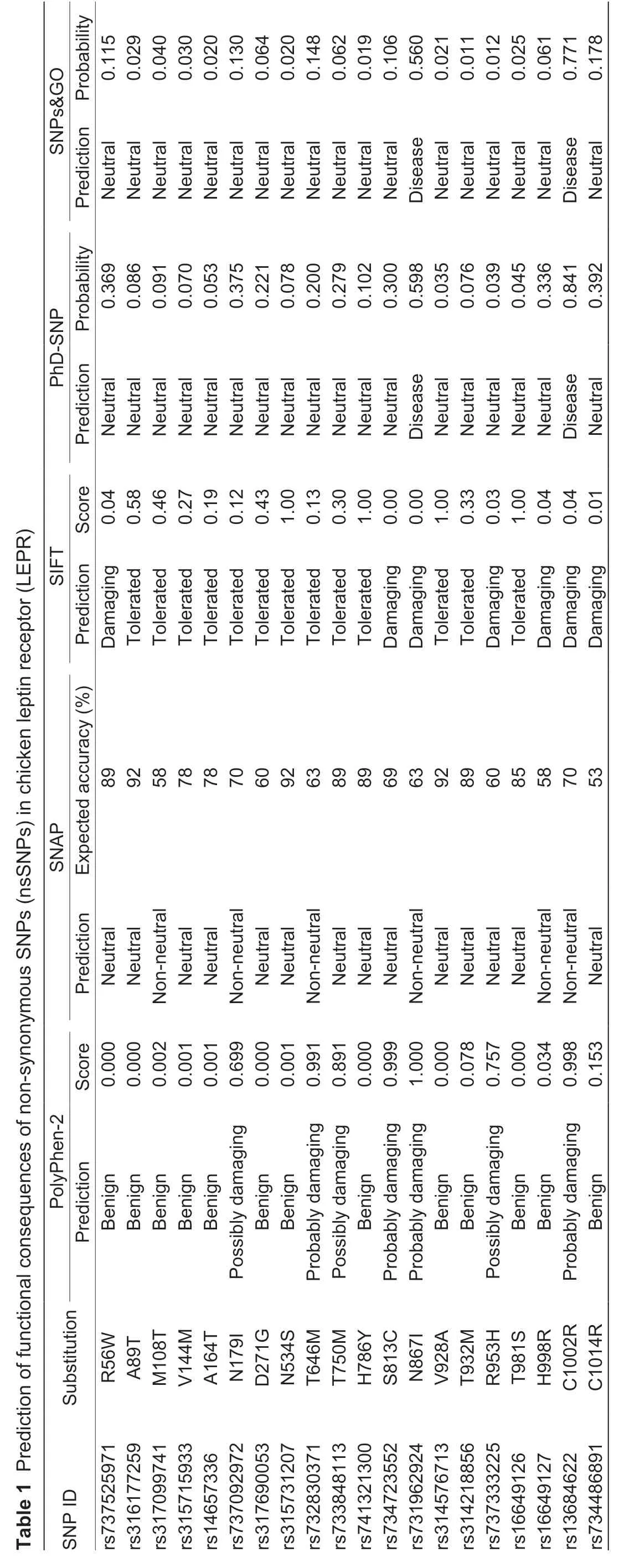
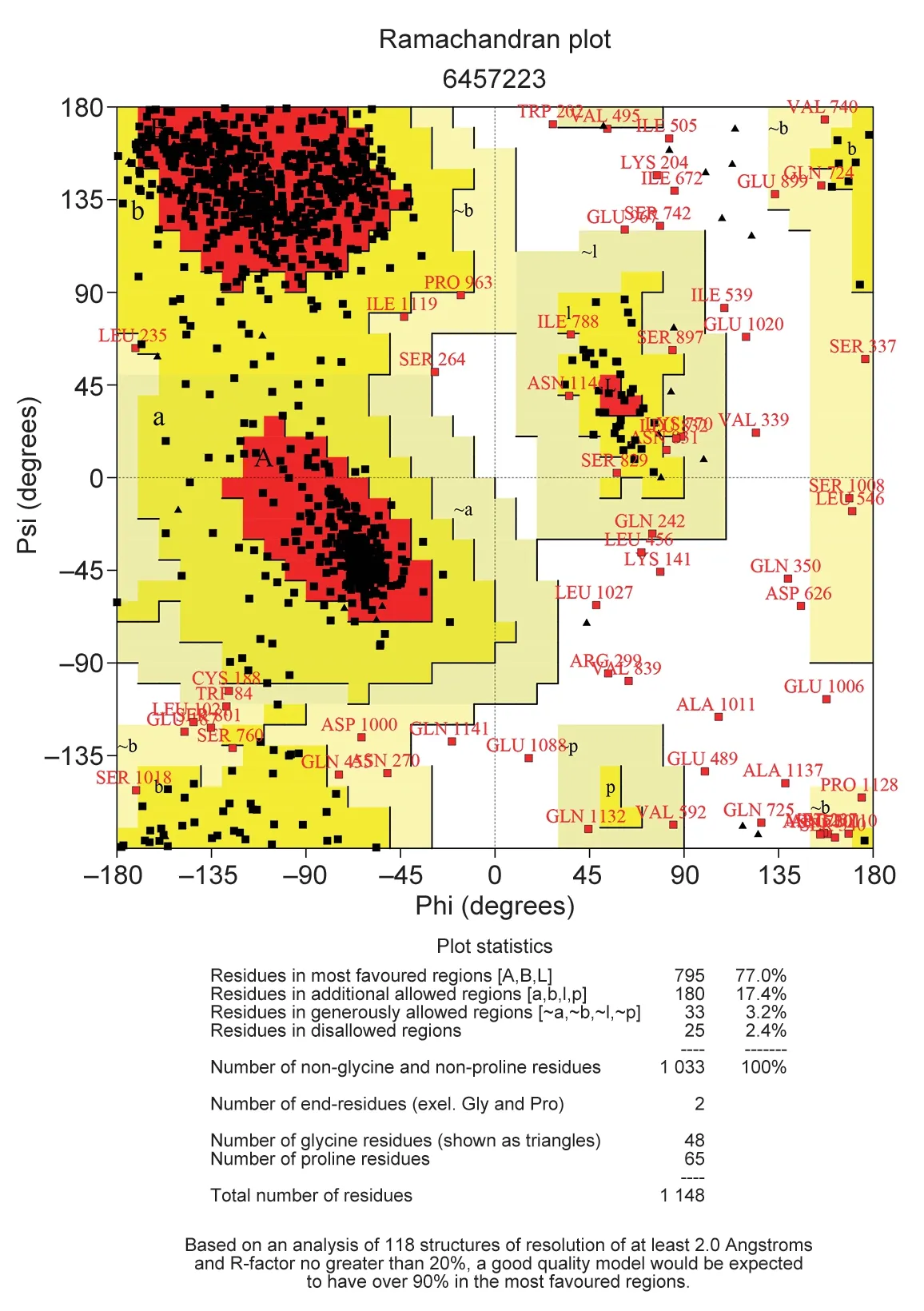
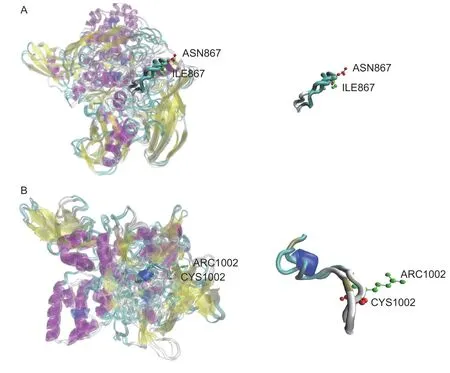
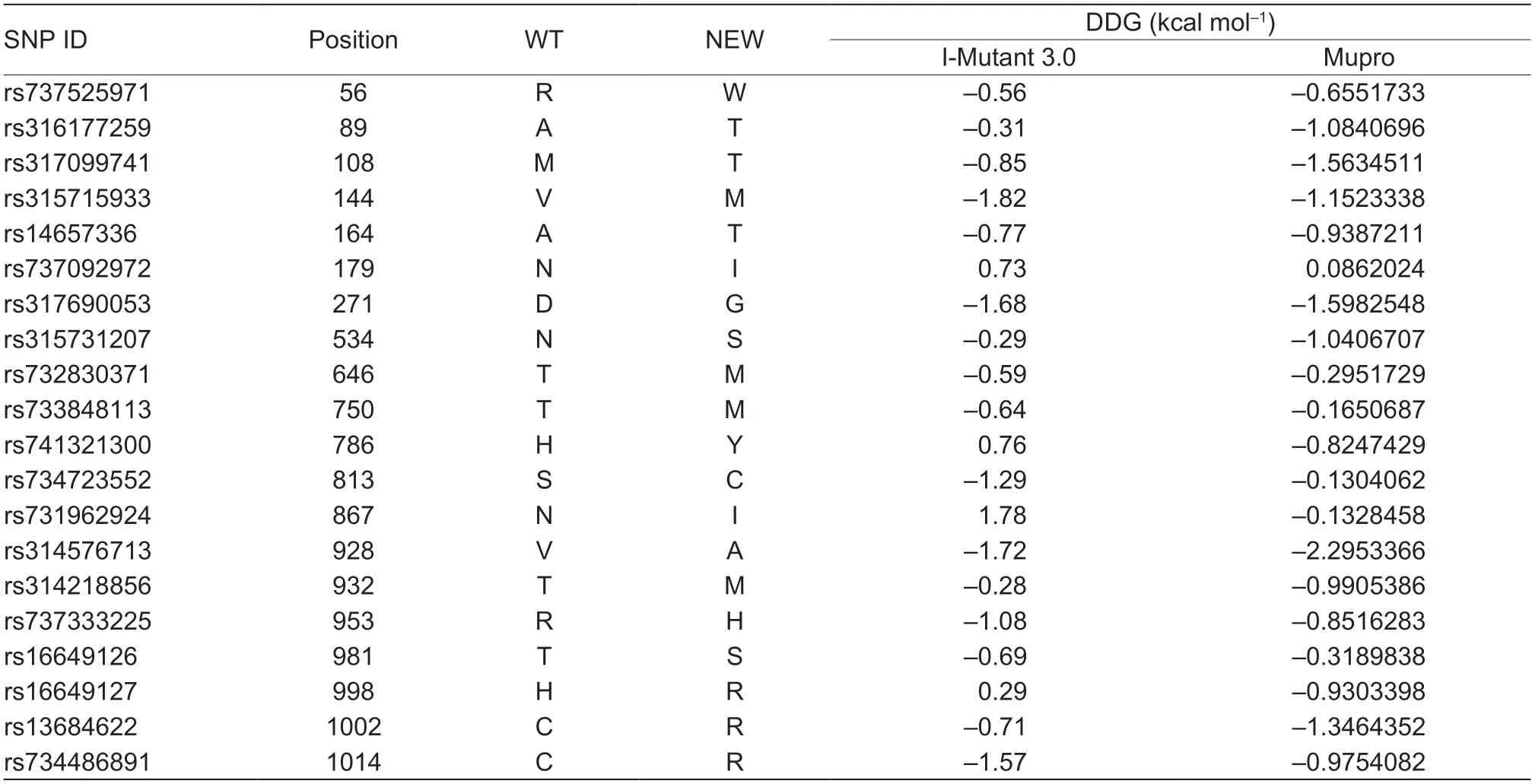
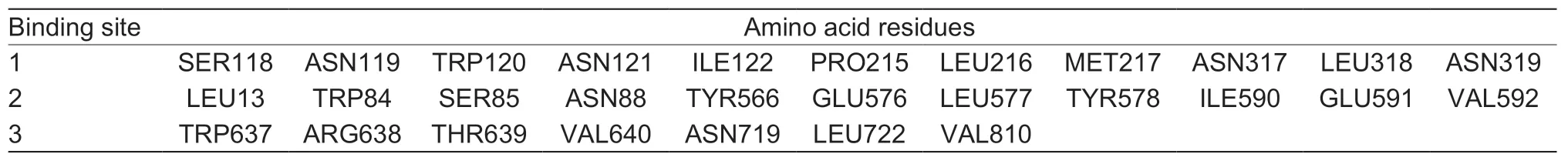
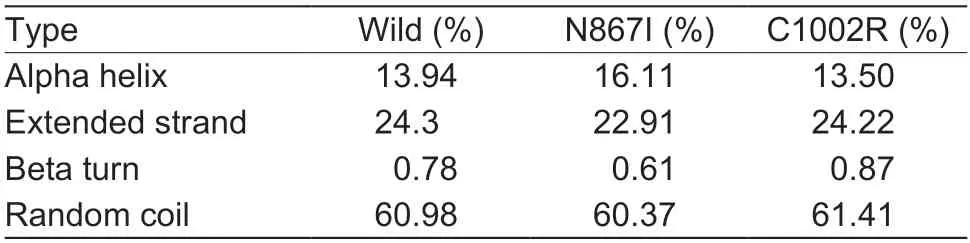
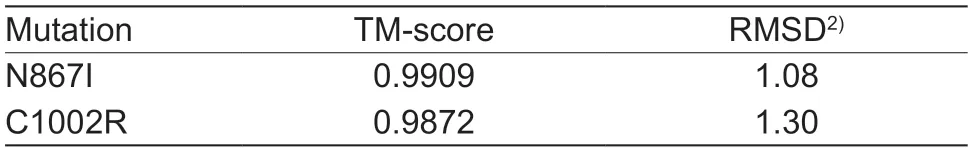
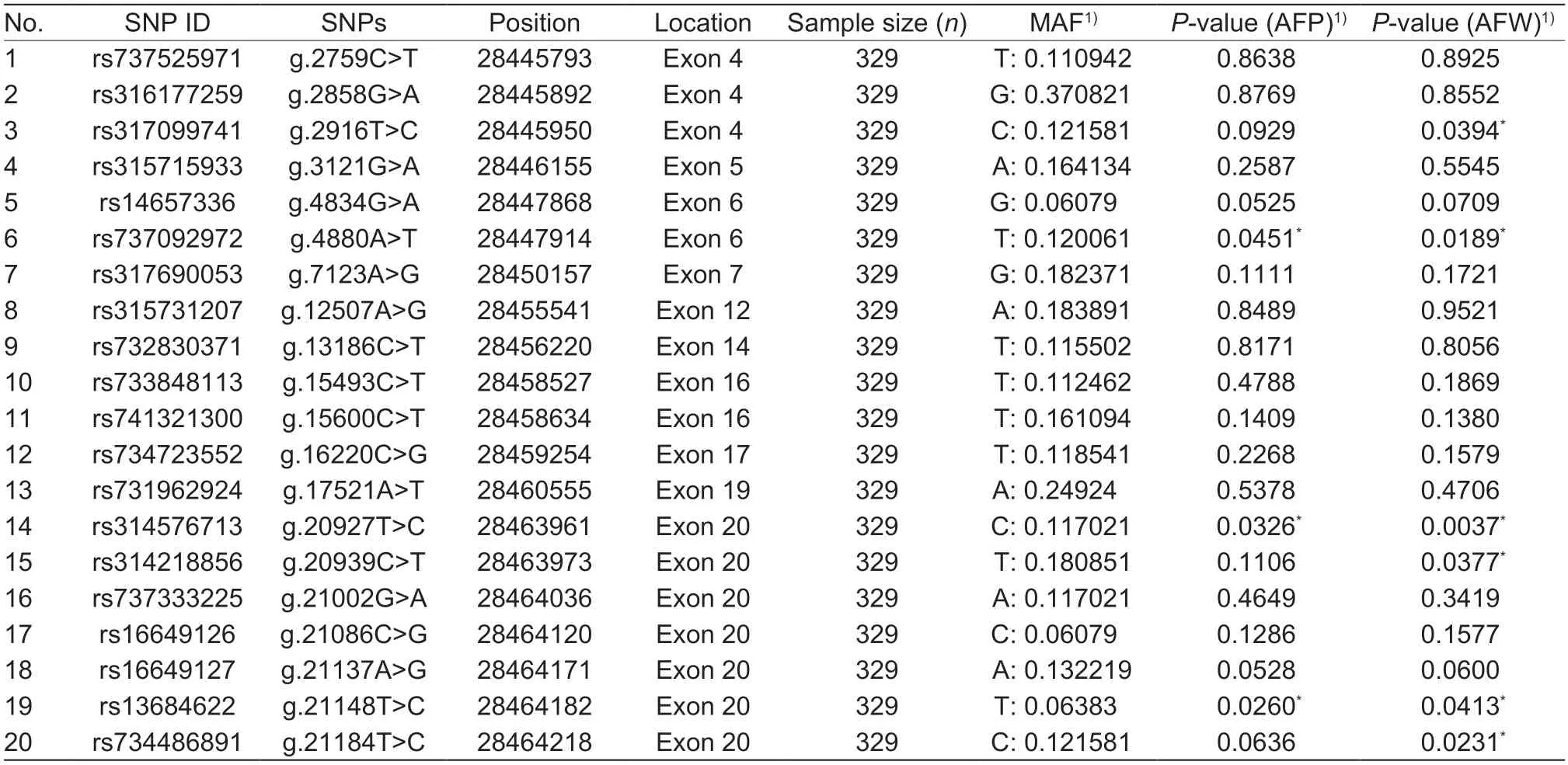
A total of 20 nsSNPs were used for the prediction of their functional effectsviaPolyPhen-2,SNAP,SIFT,PhD-SNP,and SNPs&GO tools (Table 1).Excessive fat deposition is a harmful phenotypic trait in chickens,so functional nsSNPs are also equivalent to damaging nsSNPs in this study.These nsSNPs were predicted by PolyPhen-2 to have three different types of damages:Probably-damaging(score>0.96),Possibly-damaging (0.2 According to the results of the five different software tools shown in Table 2,two amino acid mutations (N867I and C1002R) were considered to be the most likely functional nsSNPs affecting abdominal fat traits in chickens.We used the Venn Diagram package in R language to draw the Venn diagram (Fig.2). Fig.2 Venn diagram of intersection of non-synonymous SNPs(nsSNPs) data in leptin receptor (LEPR) coding region.Two SNPs,rs731962924 (N867I) and rs13684622 (N1002R),were predicted to be the most likely functional nsSNPs. We predicted the stability alteration of single-site mutations of LEPR protein by I-Mutant 3.0 and Mupro (Kamaraj and Purohit 2013;Yakubuet al.2017;AI-Shuhaibet al.2018).The results showed that most nsSNPs decreased the stability of LEPR protein,while N179I,H786Y,N867I,and H998R were considered to increase the stability of LEPR protein by I-Mutant 3.0,and N179I was considered to increase the stability of LEPR protein by Mupro (Table 2). Protein BLAST in NCBI website was used to retrieve theLEPRamino acid sequences of five other highlyhomologous species,includingSus scrofa,Homo sapiens,Rattus norvegicus,Macaca mulatta,andMus musculus.Then Clustal-Omega online website was used for multiple sequence alignment (Liet al.2018).It could be concluded that amino acid N867and amino acid C1002were both relatively conserved in the evolutionary process(Fig.3).The conserved parts of the amino acid residues of LEPR protein were calculated using ConSurf web server.We only enumerated the conserved residues that matched the two high-risk nsSNP positions we identified (Table 3).Fig.4 showed the structure,function,and conservative results of the two high-risk nsSNPs in ConSurf.According to the prediction results of ConSurf,the prediction result of amino acid N867was consistent with that of multiple alignments (Fig.3).In contrast,the prediction result of amino acid C1002was“Average”which was different from that of multiple sequence alignment. Fig.3 Highly homologous alignment of chicken leptin receptor (LEPR) protein and other species.The lower three solid histograms from top to bottom were the conservatism of multi-sequence alignment,contrast quality,and consensus sequence.The color was adjusted and edited by Jalview Software. Fig.4 ConSurf analysis was performed on two high-risk non-synonymous SNPs (nsSNPs). The MutPred2 Web Tool can predict disease-associated phenotype and also identify the molecular cause of disease that results from amino acid substitution instigated by nsSNPs.Combinations of highgscores and lowPscores were referred to as valid hypotheses,withg>0.5 andP<0.05 were referred to as actionable hypotheses,withg>0.75 andP<0.05 were referred to as confident hypotheses.The variation of N867I (g=0.517,P=0.03) and C1002R (g=0.605,P=0.03) was related to“Loss of B-factor”and“Loss of Loop”respectively (Table 4),and the confidence degree was actionable hypotheses. We used FTSite server to predict whether the identified nsSNPs were present in the LEPR protein binding region.We found three different binding sites (mesh loops) with different amino acid residues in the LEPR protein (Table 5).Previously identified possible functional nsSNPs,rs731962924 (N867I)and rs13684622 (C1002R),were predicted not in the amino acid residues that participated in the binding sites above. The changes in the secondary structure of LEPR protein caused by two high-risk site mutations were analyzed through the online website of SOPMA.Table 6 shows the average percentage of the total secondary structure content of the three protein systems on the trajectory,indicating the slight difference between wild-type and mutation-type systems. We used the MODELLERv9.23 Software to predict the 3D models of wild-type and mutant-type ofLEPRproteins.Structural homology modeling of LEPR proteins referred to four known protein structures in PDB database (Table 7).Only percent identity>30% can be used for homologous modeling.The high-resolution structure refinement and energy minimization of the atomic-level protein model was carried out by using the ModRefiner online tool in the I-TASSER website.Then the Ramachandran plot diagram (Fig.5) was drawn with PROCHECKv3.5 online tool to check the quality of the model.Most of the amino acid residues(94.4%) were in the allowable region,accounting for 77.0% (795 amino acid residues) in the most favorable region and 17.4% (180 amino acid residues) in the additional allowable region,so it could be considered that the quality of the model was of fair quality and could be analyzed later.Then we used the 3D Visualization Software VMD to observe the changes of the protein structure from a mutant with native.It can be observed that when ASN is mutated to ILE (Fig.6-A),a part of Turn is missing near the mutation site,and thereis a slight change in the morphological characteristics of the secondary structure.The secondary structures of the two proteins are not highly overlapped near the mutation site when CYS mutates to ARG (Fig.6-B),part of the 3-10-Helix structure and part of the Extended-Beta structure are missing near the mutation site,which shows that the mutation site has a significant impact on the local structure of LEPR protein.Finally,the TM-align online network tool was used to compare the RMSD value and TM-score value of the modeled mutant proteins with the wild-type (Table 8)and come to the conclusion that the two mutation sites cause local structural changes in the LEPR protein as a whole. Fig.5 Ramachandran plot of constructed leptin receptor (LEPR) protein.Most of the amino acid residues were in the allowed region. Fig.6 The structure of the mutant protein was superimposed on the wild-type leptin receptor (LEPR) protein.A,asparagine (ASN)is mutated to isoleucine (ILE).B,cysteine (CYS) is mutated to arginine (ARG).Corresponding colors represent different secondary structures.Alpha helix,purple;3-10-helix,blue;Pi-helix,red;extended-beta,yellow;bridge-beta,tan;turn,cyan;coil,white. Table 2 I-Mutant 3.0 and Mupro analysis of protein’s stability change upon amino acid substitution1) To determine which SNP(s) can affect chicken adipose deposit,we analyzed the association between genotypes of 20 nsSNPs in the coding region and abdominal fat traits.We found that three SNPs were significantly correlated with both abdominal fat weight and abdominal fat percentage (P<0.05),which were rs737092972(g.4880A>T),rs314576713 (g.20927T>C),and rs13684622(g.21148T>C),respectively (Table 9).Multiple comparison revealed that birds with genotype TT for rs737092972,CC for rs314576713,and CC for rs13684622 had more AFW and AFP than birds with their other genotypes (Appendix A). Table 3 Conservative scores of two high-risk non-synonymous SNPs (nsSNPs) using ConSurf The hypothesis that there would be functional SNPs,in exons of chickenLEPRgene in relation to AFW and AFP was supported by the results of the present study.In this study,we used various computer-based functional prediction methods to preliminarily screen the functional nsSNPs in the exon region of chickenLEPRgene.Finally,we identified two underlying functional SNP (rs731962924 (N867I)and rs13684622 (C1002R)) on the exon ofLEPRgene.Association analysis showed that rs13684622 (C1002R)was significantly associated with AFW and AFP in chickens and potentially applied to improvement of broiler abdominal fat in MAS program.Our study provides a reference for carrying out corresponding cell experiments or other in vitro experiments in the future. Studies have shown that the allelic variation of genes may be of potential importance to the genetic improvement of animals (Moreiraet al.2018).The identification of functional SNP responsible for traits of interest in domesticated animals will provide reliable molecular markers for animal molecular breeding (Ewuolaet al.2018).Given that the change of single base in the coding region of LEPR protein can lead to the change of amino acid,and affect the structure and function of the protein,here we focus on the effects of nsSNPs on the function ofLEPR.The results suggested that some of nsSNPs were predicted to be damaging and associated with phenotypic traits (AFW,AFP),whereas others were considered neutral.Integration of SNP-trait association andin-silicoanalysis can make functional SNPs identified more reliable,and offer candidates for subsequent functional validation experiments.Additionally,a combination of association and computer electronic predictive analysis provides an alternative avenue to identify important molecular markers for breeders. The interpretation of important phenotypic variations in production practice is still challenging because some SNPs may not have significant functional effects,or the frequency is very low (Arifuzzamanet al.2020).Besides,it is costly and time-consuming to identify nsSNPs related to a specific phenotype through traditional molecular experimental methods in a large population size.In this case,in-silicoanalysis approach can reveal the biological consequences of these SNPs related to the structure and function of proteins more effectively.Beforein vivoexperiments,computational tools can be effectively used to screen the related functional consequences of a large number of SNP.In order to make the prediction results more accurate and reliable,we selected five frequently used bioinformatics tools (PolyPhen-2,SNAP,PhD-SNP,SNPs&GO and SIFT) to predict the function of nsSNPs in the exon region of chickenLEPRgene,and finally determined that rs731962924 (N867I) and rs13684622 (C1002R) were identified as possible functional nsSNPs (Fig.2). Protein stability plays an important role in normal biological function,activity and regulation.I-Mutant3.0 and Mupro were used to predict the change of protein stability based on unit point mutation.The change of protein stability is usually accompanied by the change of free energy (ΔG).The results showed that most of the amino acid substitutions were highly unstable (Table 2).These single amino acid mutations can cause abnormal protein folding,changes in main chain tension and electrostatic force,which in turn leads to the increase of protein aggregation or degradation(Wanget al.2020). Table 4 Prediction of disease-related amino acid substitution and phenotypes by MutPred2 Table 5 Analysis of ligand binding site with FTSite Server Table 6 Percentage of the secondary structure of wild type and mutant type It has been found that conserved sequences often correspond to important functional regions,and nsSNPs located at highly conserved sites are more likely to become functional SNP than non-conserved nsSNPs.Multiple sequence alignment among different species and ConSurf conservation estimation can be used to screen conserved sites of amino acid sequences,and screen possible functional nsSNPs.It was confirmed that N867was a conservative amino acid site,while C1002was highly conserved in the results of multiple sequence alignment,and ConSurf analysis showed“Average”(Fig.3;Table 3).We believe that the difference between the two results occurs due to the poor conservation of amino acids around the amino acid C1002,which affects the conservation of C1002amino acids,or it may be caused by the lack of species used in multiple sequence alignment (Pollastriet al.2002;Ezawa 2016). Table 7 Four known protein structures used in leptin receptor (LEPR) protein modeling Table 8 The TM-align result after superposition of leptin receptor (LEPR) protein mutant and wild type1) SinceLEPRgene is a high-affinity receptor for leptin,we used online bioinformatics tools to study the effect of nsSNPs on LEPR protein binding sites.The predictionof ligand binding sites has a wide range of applications,including structure-based functional prediction and explanation of the functional relationship between receptors and ligands among different proteins (Nganet al.2012).In this study,the amino acid substitution caused by the two functional nsSNPs identified was not involved in the amino acid residues of the three binding sites.So,we believe that these two nsSNPs may not cause changes in LEPR protein binding sites. We used MutPred2 online website to predict the possible pathogenic molecular mechanism of two highly pathogenic nsSNPs.The consequence of N867I is“Loss of B-factor”.The B-factor,also known as the“Temperature Factor”is an indicator of a protein’s static flexibility,and the loss of the B-factor may mean that the mutation leads to a decrease in the thermal stability of the LEPR protein (Duanet al.2016).The consequence caused by C1002R is“Loss of Loop”.Proteins use the conformational variability of the Loop region to perform different biological tasks,including molecular recognition and signal transduction.Loss of Loop means that the mutation may cause theLEPRbarrier when binding to leptin,thus affecting the information transmission inside and outside the cell (Mandellet al.2009). The structural model of proteins helps us to understand biological processes at the molecular level (Wiltgen 2009).Local conformational changes,such as conformational changes related to binding sites,may have greater negative effects on protein function,resulting in changes in pathogenicity or important animal traits.For example,recently,Dakalet al.(2017) showed that some SNPs in human IL-8 proteins caused local changes in the structure–function relationship,which might be associated with the occurrence of diseases such as cancer.These experimental evidences support that intricate three-dimensional structural details are essential for protein function.To further investigate how these mutations affect the structure and function of proteins,we analyzed the overlapping structures of wild-type and mutant (N867I and C1002R) proteins(shown in Fig.6).The effect of nsSNPs on protein structure and function can be better understood by mapping nsSNPs to the corresponding three-dimensional structure of the protein.Based on four known protein structures (Table 7),the LPER tertiary structure of wild-type mutant protein was modeled by MODELLERv9.23 Software so as to infer possible structural-functional consequences of nsSNPs in LEPR protein.Four known protein structures were used simultaneously,which would make the model constructed in this study more accurate than traditional methods (Webb and Sali 2014a,b;Al-Shuhaibet al.2018).With ModRefiner to further refine and minimize energy,the goal of energy minimization is to model protein structures without space collisions and potential energy to obtain the most stable structure of thermodynamically.To extend our structural analysis,TM-score and RMSD were used to evaluate the topological similarity between wild-type and mutant models and the deviation of mutant structures from their original configuration (Li 2013).The above results showed that the two identified nsSNPs could affect the protein structure in the mutation location,while the effect of C1002R on protein structure was more significant. Table 9 Association of 20 non-synonymous SNPs (nsSNPs) in exon of leptin receptor (LEPR) gene with AFW and AFP Association analysis indicated that there were three SNPs (rs737092972,rs314576713 and rs13684622) with significant effects on AFW and AFP.However,they differed greatly in the number of genotypes (Appendix A).This could be attributed to divergent selection for abdominal fat percentage of NEAUHLF,thereby leading to great changes of allele frequencies of these SNPs inLEPRgene,which in turn supports thatLEPRgene could play a vital role in chicken adipose deposition.The research work has brought about a discovery of consequences of the rs13684622(C1002R) in the structural and functional proprieties of the chicken LEPR protein using computational biology tools,which is in accordance with a significant association of the rs13684622 (C1002R) with AFW and AFP in chickens.The homozygous genotype (CC) of rs13684622 (C1002R) can significantly increase AFW and AFP of chicken compared with the heterozygous genotype (TC),indicating that homozygous genotype (CC) is unfavorable genotype for reducing abdominal fat.The findings may have a practical application in chicken breeding program. Taken together,by using integration of computer-based functional prediction and SNPs-traits association analysis,we hold that rs13684622 (C1002R) mutant ofLEPRmay be an important functional SNP affecting chicken abdominal fat deposition,and promisingly applied to improvement of broiler abdominal fat in future MAS program.Also,it is necessary that further functional experiments are designed to verify effects of this SNP onLEPR. Acknowledgements This work was supported by the National Natural Science Foundation of China (31572394),the China Agriculture Research System of MOF and MARA (CARS-41),and the White Feather Broiler Breeding Joint Project of the Ministry of Agriculture and Rural Affairs of China (19190526). Declaration of competing interest The authors declare that they have no conflict of interest. Ethical approval All studies involving animals were conducted according to the guidelines for the Care and Use of Experimental Animals established by the Ministry of Science and Technology of the People’s Republic of China (approval number 2006-398).This study was approved by the Laboratory Animal Management Committee of Northeast Agricultural University,China. Appendixassociated with this paper is available on http://www.ChinaAgriSci.com/V2/En/appendix.htm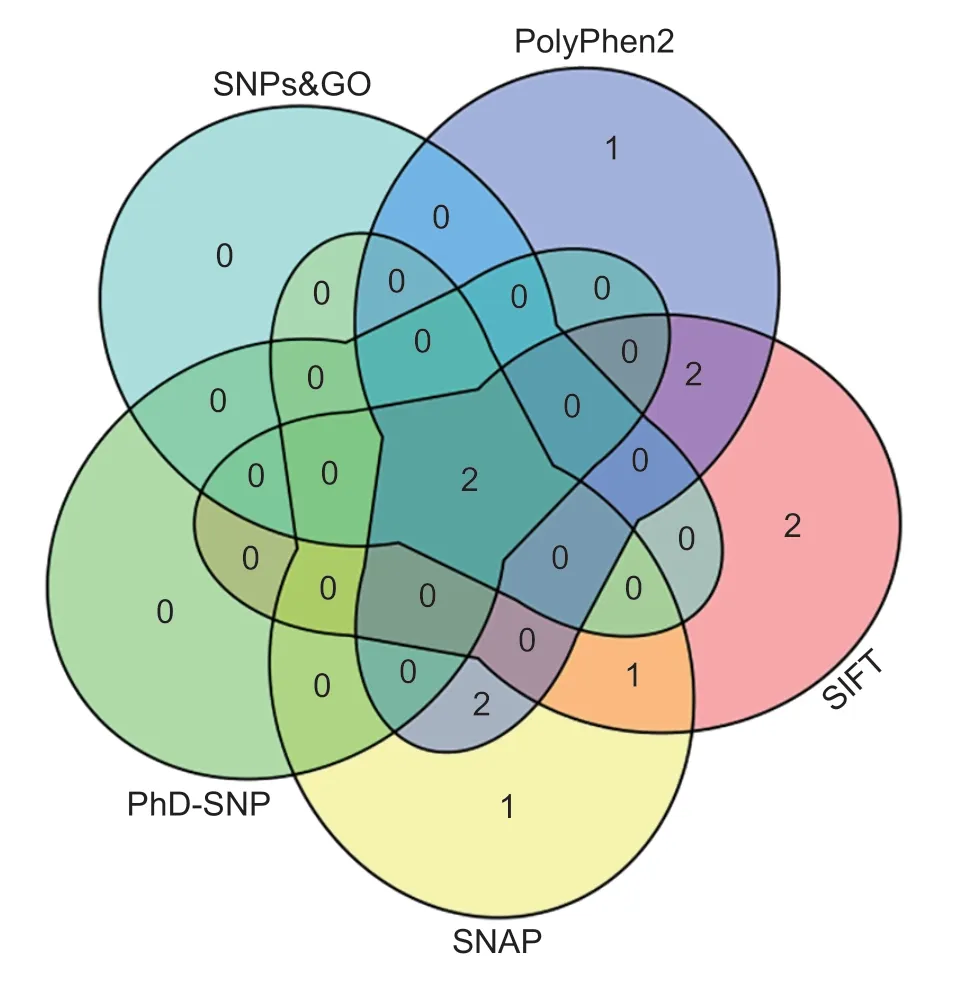
3.3.Analysis of nsSNPs based on stability
3.4.Analysis of highly conserved amino acid residues
3.5.Predicting the consequences of two of the most deleterious amino acid substitutions
3.6.Prediction of ligand binding sites of LEPR protein with FTSite
3.7.Changes of the secondary structure after mutation of LEPR protein
3.8.Structural modeling and model quality inspection
3.9.Association between nsSNPs and abdominal fat traits in chickens

4.Discussion


5.Conclusion
杂志排行

Journal of Integrative Agriculture的其它文章
- Assessing the impact of non-governmental organization’s extension programs on sustainable cocoa production and household income in Ghana
- Triple bottom-line consideration of sustainable plant disease management:From economic,sociological and ecological perspectives
- A rice geranylgeranyl reductase is essential for chloroplast development
- lmpacts of climate change on drought risk of winter wheat in the North China Plain
- Rapid determination of leaf water content for monitoring waterlogging in winter wheat based on hyperspectral parameters
- Does nitrogen application rate affect the moisture content of corn grains?