大豆分离蛋白/魔芋葡甘聚糖复合脂肪模拟物的制备及结构分析
2021-08-09雷爱玲范盛玉王亚楠陈厚荣张甫生
雷爱玲,范盛玉,王亚楠,陈厚荣,张甫生
(西南大学 食品科学学院,重庆,400715)
超重和肥胖已成为我国乃至全球严重的公共卫生问题[1],消费者对健康低脂产品具有很高的需求,消费低脂类产品已成为一种生活方式[2]。如何在降低脂肪摄入量的同时不影响食品的风味和口感成为食品领域的研究热点,基于此脂肪模拟物应运而生。大豆分离蛋白(soy protein isolate,SPI)是应用最广泛的植物蛋白,其蛋白质含量在90%以上,具有胶凝、乳化、溶解、起泡、成膜等多种优良特性,还能加快脂肪与能量的新陈代谢,与多糖混合可提供一种类似于脂肪的味道[3],现已有SPI在低脂食品方面的研究。PAGLARINI等[4]利用SPI制备乳液凝胶替代猪背脂肪,发现替代物不影响法兰克香肠的质地、流变和感官性能。SUN等[3]发现SPI和纳米纤维素比例为7∶1时,作为脂肪替代物加入冰淇淋中,取代10%的奶油,其接受度最高。
魔芋葡甘聚糖(konjac glucomannan,KGM)是从魔芋块茎中提取的一种天然高分子中性杂多糖[5],由D-甘露糖和D-葡萄糖通过β-1,4糖苷键聚合而成,其在C-6位置有一个低程度的乙酰基,在C-3位置有一些分支,这决定了它在水中的溶解度。其吸水性强、膨胀率高、黏度大且成膜性能极强,已被证实对人体健康有显著的益处,能有效抑制脂肪酸的合成,具有一定的减肥效果。此外,KGM还具有抗高血糖和高胆固醇血症、抗炎症、益生元活性、预防癌症等[6]功能。正是这些特性使KGM有潜力作为脂肪模拟物应用到食品中。DAI等[7]用KGM作为脂肪替代品应用于马苏里拉奶酪中,发现KGM能提升马苏里拉奶酪的一些理化特性。LI等[8]将微粒化魔芋凝胶作为蛋黄酱中的脂肪模拟物,制得的低脂蛋黄酱具有更低的热量和更高的贮藏稳定性,然而脂肪模拟物的添加对蛋黄酱的外观和颜色有不利影响,替代水平低于30%时,可被接受。
目前,菊糖[9]、KGM[7]、乳清蛋白[10]、改性淀粉[11]等单一碳水化合物或蛋白质已作为脂肪模拟物应用于食品中,但由于单一脂肪模拟物没有较好的贮藏稳定性,限制了其在食品中的应用。植物蛋白和多糖是2种重要的天然高分子生物聚合物,也是影响食品风味和质地的主要成分,二者相互作用会改变体系的结构、质地甚至产生新的功能特性,研究表明蛋白多糖复合体系可改善产品的表面活性[12]、乳化性、质地特性[13]并增强其凝胶性[14]。而目前蛋白-多糖复合物作为脂肪模拟物还有待进一步的研究。基于此,本研究以SPI和KGM为研究对象,通过湿热处理制备复合脂肪模拟物,并探究总固形物含量、KGM添加量、加热温度、加热时间对其性能的影响,通过响应面试验优化复合脂肪模拟物的制备工艺,最后观察复合脂肪模拟物制备工艺过程中粒径颗粒分布及微观结构的变化,从而得到润滑、细腻的脂肪模拟物,以期为复合脂肪模拟物的深入研究提供一定的理论依据。
1 材料与方法
1.1 试验材料
大豆分离蛋白(食品级),蛋白质含量>90%,上海源叶生物科技有限公司;魔芋胶(食品级),葡甘聚糖含量≥92%,上海北连生物科技有限公司。
1.2 仪器与设备
DF-101S型集热式恒温加热磁力搅拌器、HH-ZK8型数显恒温水浴锅,巩义市予华仪器有限责任公司;MCR302型模块化旋转与界面流变仪,奥地利安东帕有限公司;78-1型磁力加热搅拌器,常州溴华仪器有限公司;XHF-D型高速分散器、SCIENTZ-10 ND型真空冷冻干燥机,宁波新芝生物科技股份有限公司;METTLER TOLEDO型高精数显电子天平,梅特勒-托利多仪器(上海)有限公司;Mastersizer 3000激光粒度仪,英国马尔文仪器有限公司;Phenom Pro型扫描电镜,荷兰Phenom World公司。
1.3 试验方法
1.3.1 SPI/KGM复合脂肪模拟物的制备
称取一定量的SPI溶解于蒸馏水中,在室温下磁力搅拌1 h,待SPI完全溶解形成SPI悬浮液,向其中慢慢加入已称量好的KGM,使二者充分搅拌均匀。然后置于恒温水浴锅中进行湿热处理,使蛋白适度变性,将湿热处理后的复合物冷却至室温,于4 ℃冰箱放置过夜,使其完全水化,以形成稳定、柔软的复合体系。最后使用高速剪切机作微粒化处理即可得SPI/KGM复合脂肪模拟物。
1.3.2 单因素试验设计
选取总固形物含量(4%、6%、8%、10%、12%,质量分数)、KGM添加量(0.2%、0.4%、0.6%、0.8%、1.0%,质量分数)、加热温度(55、65、75、85、95 ℃)、加热时间(5、15、25、35、45 min)为试验因素,探究其对SPI/KGM复合体系膨胀率及黏度的影响。
1.3.3 响应面试验设计
在单因素试验结果的基础上,采用Box-Behnken Design的设计原理进行响应面优化。以KGM添加量、总固形物含量、加热温度作为响应因素,SPI/KGM复合体系的膨胀率和剪切速率为0.1/s时的黏度为响应值,设计3因素3水平的响应面试验,获得SPI/KGM复合脂肪模拟物的最佳制备条件,所有试验重复3次。响应面试验设计如表1所示。
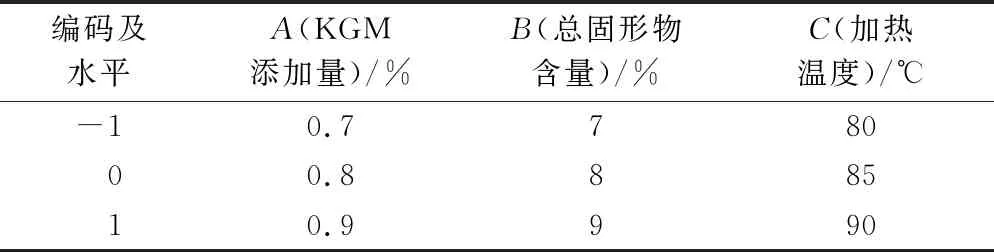
表1 响应面试验因素与水平
1.3.4 SPI/KGM复合脂肪模拟物的性能测定
1.3.4.1 膨胀率的测定
称取一定量的SPI/KGM复合体系于烧杯中,并记录空烧杯的质量m1,用打蛋器打发2 min后记录混合物的质量m2,然后在另一空烧杯m3中加入与打发后的混合物同样体积的水,并记录其质量为m4,SPI/KGM复合体系的膨胀率按公式(1)计算[15]。

(1)
1.3.4.2 黏度的测定
使用流变仪对样品进行静态剪切流变测试[16],测试条件:50 mm平行板夹具,测试间距1 mm,测试温度25 ℃,稳态模式,加样后平衡2 min,剪切速率为0.1~100/s,取点模式为对数取点,取点数30个,测定各样品黏度随剪切速率变化的情况。
1.3.5 粒径的测定
采用激光粒度仪测定样品的颗粒大小,向装有水的烧杯中缓慢加入样品,待遮光度显示8%~20%时进行测定[17]。参数设置:SPI折射率1.589,吸收率0.001,水折射率1.333,搅拌速度2 400 r/min,遮光度为8%~20%。重复测定6次,记录结果的D10、D50、D90和D[4,3]。
1.3.6 微观结构的测定
将制备好的样品倒入90 mm培养皿中,使样品均匀地涂膜在培养皿内,随后置于冰箱预冷冻24 h,使样品完全结成冰,放入真空冷冻干燥机中48 h,使样品完全干燥,待用。用固定胶将样品黏于双面导电的样品铜台上,喷金[18]。之后在300和1 000放大倍数下使用扫描电镜观察,并选取有代表性的视野进行拍摄。镀金法:真空度10-2~10-3Torr(1 Torr=133.322 Pa),溅射电压1.1~1.2 kV,镀膜时间2~3 min。镀膜后样品放置10 min。
1.4 数据统计与分析
试验数据均用Origin 2019、SPSS Statistics 25和Design-Expert 8.0.6进行处理与分析,每次试验3次平行,试验结果以“平均值±标准差”表示,显著性分析以Duncan’tests进行方差分析(ANOVA),P<0.05被认为存在显著差异。
2 结果与分析
2.1 单因素试验结果
2.1.1 KGM添加量对SPI/KGM复合体系性能的影响
黏度一般指施加于流体的应力和由此产生的变形速率以一定的关系联系起来的流体宏观属性,表现为流体的内摩擦。膨胀率是蛋白质起泡能力的表现。KGM添加量对SPI/KGM复合体系性能的影响如图1所示,KGM添加量在0.2%~1.0%时,SPI/KGM复合体系黏度一直增加,并且随着剪切速率的增加,复合体系的黏度均呈下降趋势,表现出典型的剪切变稀的非牛顿流体特征[19]。KGM添加量为0.2%~0.8%时,其膨胀率逐渐增强,并在KGM添加量为0.8%时,膨胀率最大,为49.29%。当KGM添加量为1.0%时,其膨胀率降低8.56%。随着KGM添加量的增加,SPI和KGM表现出较好的兼容性,使得二者的相互作用增强,分子间结构发生变化,膨胀率增强,黏度增加[20]。
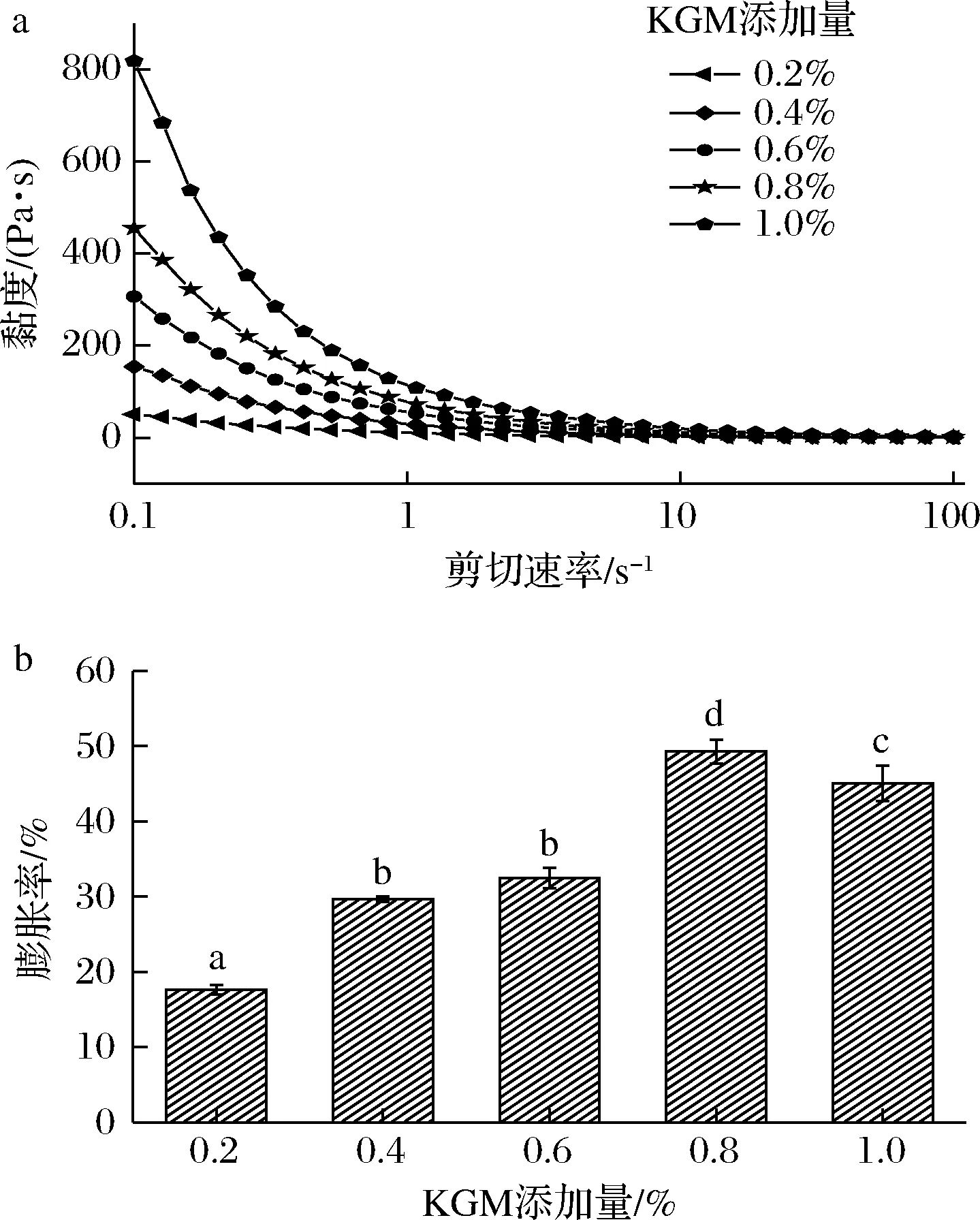
a-黏度;b-膨胀率
当KGM添加量>0.8%时,形成的复合体系硬度较大,二者的协同作用达到极点,故膨胀率降低。因此,选择KGM添加量0.8%为最佳参数。
2.1.2 总固形物含量对SPI/KGM复合体系性能的影响
如图2所示,总固形物含量在4%~12%时,随着总固形物含量增加,复合体系的黏度呈上升趋势;在4%~8%时,膨胀率随着总固形物含量的增加而增加,并在8%时达到最大值,相较于4%,其膨胀率增加了12.92%;而在10%和12%,其膨胀率较最大值分别降低了2.68%、29.67%。SPI成凝胶的临界浓度是8%,当总固形物含量为4%和6%时,二者未形成凝胶,故其黏度变化不大,表现为牛顿流体特征。而在高浓度(>6%)下,表现为牛顿流体,与田少君等[21]的研究结果一致。随着总固形物含量增加,分子间形成了更紧密的网状结构,使体系的黏度增大。此外,蛋白质在受热过程中,暴露出更多的疏水基团,增大了物质分子间相互作用力的位点[22],在高速搅打的情况下,复合体系吸收和保持空气数量的能力增强,故而膨胀率增强。当总固形物含量>10%时,所形成的复合体系黏度过高,搅打困难,故膨胀率降低。因此,选择总固形物含量为8%进行后续优化试验。
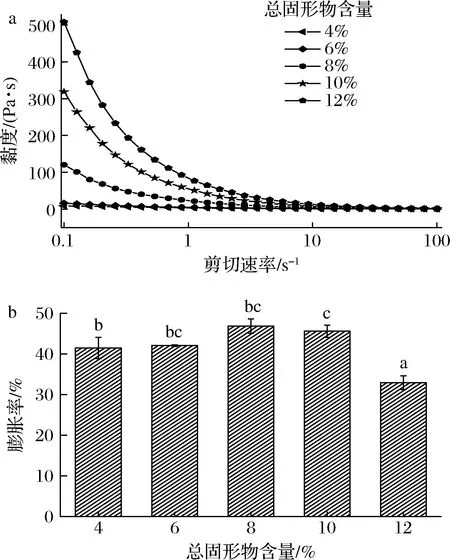
a-黏度;b-膨胀率
2.1.3 加热温度对SPI/KGM复合体系性能的影响
如图3所示,随加热温度的增加,复合体系的黏度和膨胀率呈先增加后降低的趋势。在55~85 ℃时,复合体系的黏度和膨胀率随加热温度的增加而增加,且在85 ℃时达到极大值358.3 Pa·s;当加热温度为95 ℃,与85 ℃相比黏度降低了39.44%,膨胀率降低了7.56%。与此同时,在不同加热温度下,复合体系的黏度均随剪切速率的增大而减小。究其原因可能是温度的增加,蛋白质分子呈舒展状态,原来包埋在卷曲结构内部的疏水基团暴露在外面,与多糖发生相互作用,分子间发生聚集[23],导致复合体系黏度增加。但较高的温度会破坏蛋白质的内部结构,使得体系内分子间的相互作用减弱,从而黏度下降。当温度为55 ℃时,蛋白质开始变性,两分子间还未出现交联,聚集现象尚未表露,结合较弱[24]。因此,搅打下吸入空气的能力较弱,膨胀率较弱。随着温度的升高,分子间的作用力增强,内部结合更紧密,故膨胀率增强。而当温度继续升高至95 ℃时,蛋白质分子中氢键、疏水作用等非共价键被破坏,使得分子间结合能力减弱,膨胀率降低。综合考虑,选择加热温度为85 ℃作为下一步试验的最优参数。
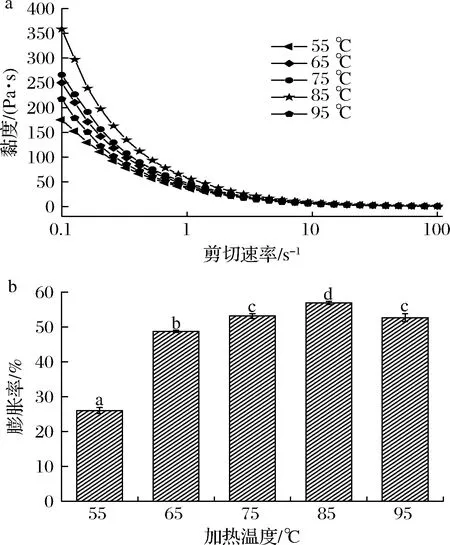
a-黏度;b-膨胀率
2.1.4 加热时间对SPI/KGM复合体系性能的影响
如图4所示,随加热时间的延长,复合体系的黏度呈现先增加后减小的变化。在5~25 min时,随加热时间的延长,复合凝胶的黏度一直处于增大趋势,并在25 min达到黏度最大值337.8 Pa·s,可能是在加热过程中,分子结构被逐渐打开,结合了更多的水分[25]。但加热时间>25 min时,其黏度下降,到45 min时,降至254.8 Pa·s,这可能是由于长时间加热会破坏蛋白-多糖分子间的相互作用以及与水分子间的结合[26],使复合体系变得松散,导致其黏度下降。复合体系的膨胀率随加热时间的延长先增加后基本趋于稳定。在5~35 min时,其膨胀率增加了55.56%,其原因在于加热时间延长,蛋白质表面疏水基团逐渐暴露,与多糖结合形成了更稳定的复合结构,从而膨胀率增加。综合考虑,固定加热时间为25 min。
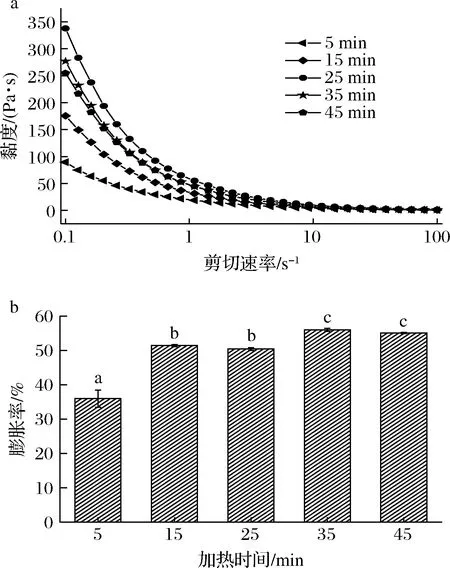
a-黏度;b-膨胀率
2.2 响应面试验结果
2.2.1 响应面试验设计及结果
响应面分析是设计试验的一种数学和统计方法,它能使试验的次数最小化,以满足特定数量的因素及其水平按试验设计进行试验[27-28]。综合单因素试验结果,以总固形物含量、KGM添加量、加热温度为响应因素,SPI/KGM复合体系膨胀率和黏度为响应值进行Box-Behnken响应面优化试验。试验设计及结果见表2。
2.2.2 响应面方差分析结果
利用Design-Expert 8.0.6软件对表2试验结果进行二次多元回归拟合,回归模型的方差分析结果见表3。模型P<0.01为极显著,失拟项P>0.05为不显著,说明建立的回归方程拟合度较高。
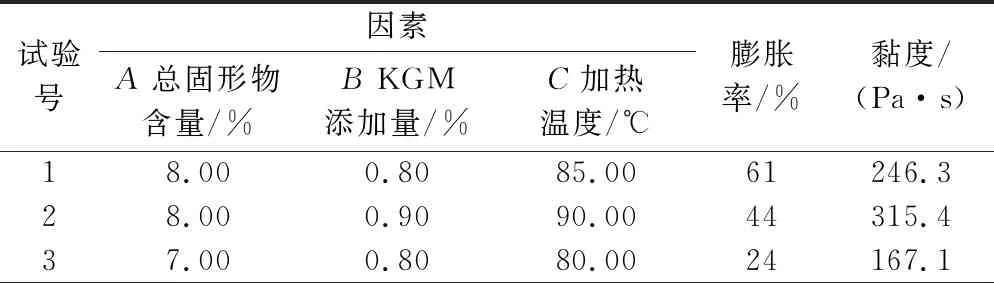
表2 响应面试验设计及结果
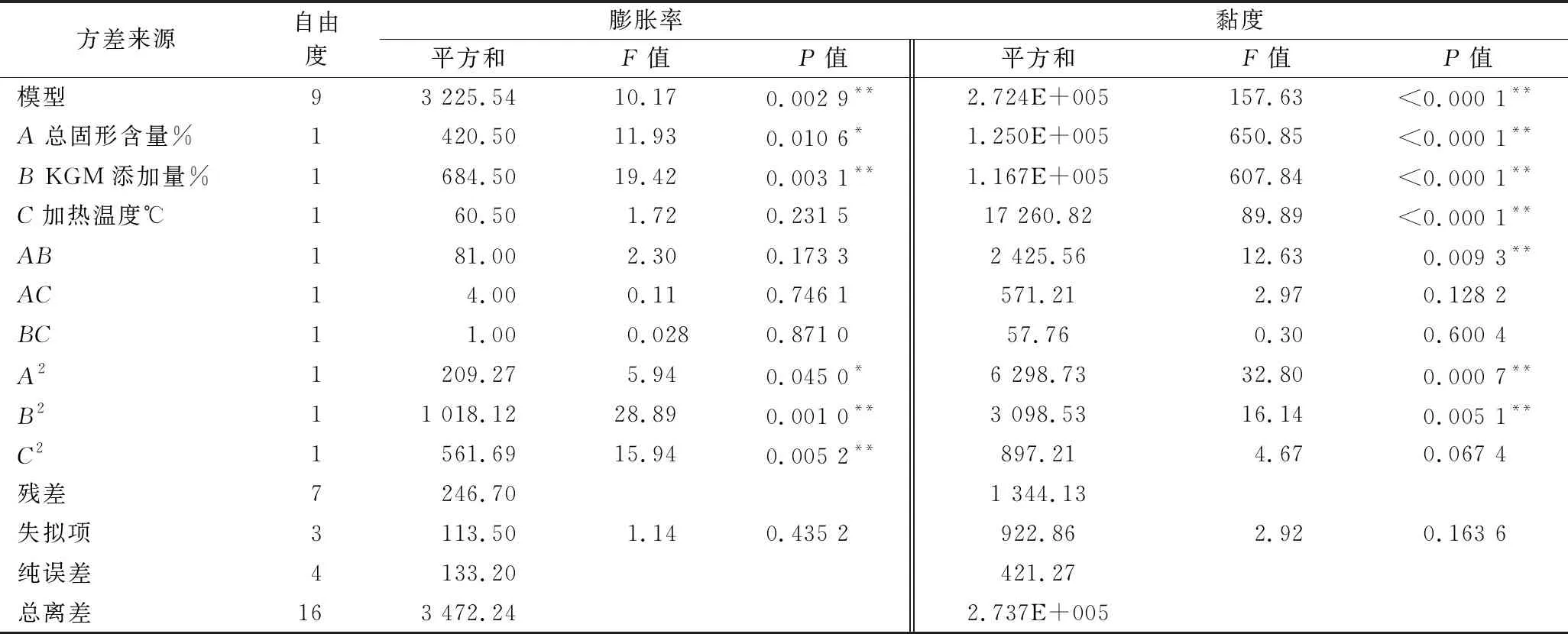
表3 回归方程方差分析
对于膨胀率模型,其拟合回归方程为:
Y1=0.55+0.073A+0.092B+0.028C+0.045AB-0.010AC+5.000E-003BC-0.071A2-0.16B2-0.12C2,R2=0.929 0
3种因素对SPI/KGM复合体系膨胀率影响程度的大小顺序为:KGM添加量>总固形物含量>加热温度。一次项B与二次项B2、C2影响极显著(P<0.01),一次项A与二次项A2影响显著(P<0.05)。
对于黏度模型,拟合回归方程为:
Y2=235.62+124.99A+120.79B-46.45C+24.62AB-11.95AC-3.80BC+38.68A2+27.13B2-14.6C2,R2=0.995 1
3种因素对SPI/KGM复合体系黏度大小的影响程度为:总固形物含量>KGM添加量>加热温度。一次项A、B、C和二次项A2、B2以及交互项AB影响极显著(P<0.01)。
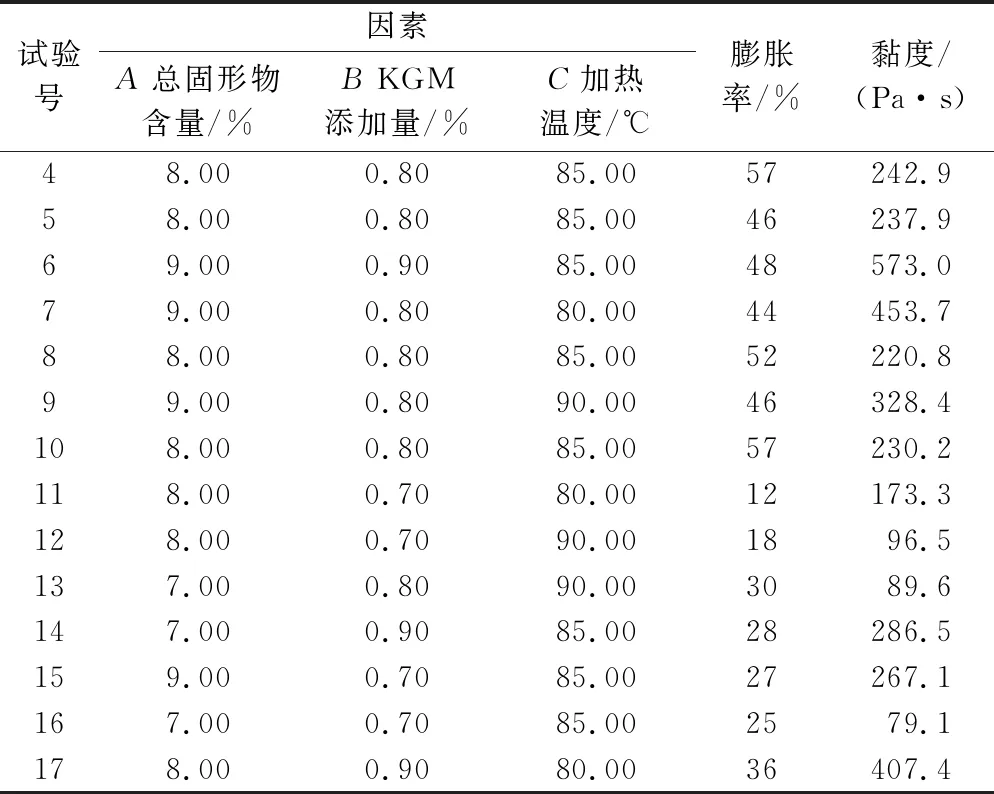
续表2
2.2.3 响应面分析
由响应面图和等高线图可直观地反映两因素间的交互作用,如图5所示,在交互项对膨胀率大小的影响中,各因素间交互作用的响应面图坡度较平缓,等高线较为松散,交互作用不显著,与方差分析结果一致。如图6所示,总固形物含量和KGM添加量交互作用的等高线较为密集,响应面图坡度较为陡峭,说明两者的交互作用显著,与方差分析结果一致。而总固形物含量和加热温度,KGM添加量和加热温度间交互作用的等高线松散,响应面图坡度平缓,说明其交互作用对SPI/KGM复合体系的黏度影响较弱。
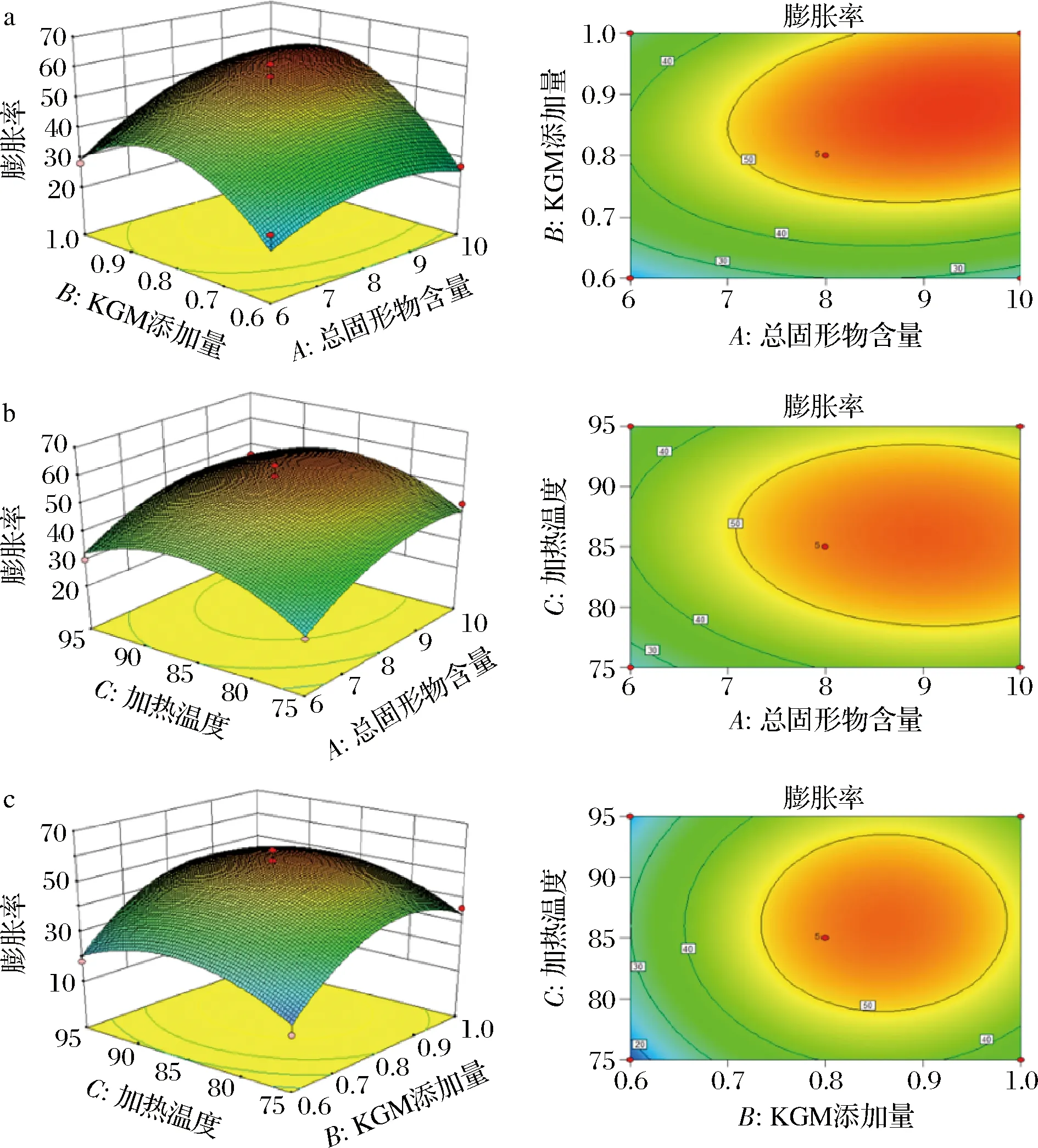
a-总固形物含量和KGM添加量;b-总固形物含量和加热温度;c-KGM添加量和加热温度

a-总固形物含量和KGM添加量;b-总固形物含量和加热温度;c-KGM添加量和加热温度
2.2.4 SPI/KGM复合体系最优工艺的确定
通过Design-Expert 8.0.6软件设计,得到的SPI/KGM复合体系的膨胀率和黏度的预测值分别为55%和237 Pa·s。考虑实际方便,调整模型最优制备工艺参数为:总固形物含量8%、KGM添加量0.8%、加热温度87 ℃。在此优化条件下对SPI/KGM复合体系进行3次平行试验,测得SPI/KGM复合体系的膨胀率为53%,黏度为233.3 Pa·s。与预测值的相对误差分别为3.64%、1.56%,均在可接受的范围内。说明利用响应面优化法得到的回归模型制备工艺参数能较准确地预测SPI/KGM复合体系膨胀率和黏度的大小,其结果真实可靠,具有重现性。
2.3 SPI/KGM复合脂肪模拟物粒径的变化
蛋白质基脂肪模拟物的制备是基于蛋白质在加热的条件下变性,使得疏水基团和区域暴露在分子表面,模拟油脂的疏水性状,然后经微粒化处理降低颗粒直径[29]。人体口腔黏膜对一定大小和性状的颗粒的感知程度有一定的阈值,当粒径<10 μm时已无法分辨出颗粒,从而能模拟出脂肪滑腻、柔软的感觉[30-31]。脂肪模拟物制备过程的粒径分布见表4和图7所示,SPI/KGM复合体系在加热前,其颗粒大小在83.4 μm左右;经热处理后,其粒径颗粒增加至105 μm左右,原因在于样品受热膨胀,颗粒明显变大;随着破碎时间的延长,粒径分布各项参数呈下降趋势。当微粒化处理20 min时,50%的样品颗粒直径在8.75 μm左右,符合以蛋白质为基质制备脂肪模拟物的颗粒要求(<10 μm)。此外,破碎时间越长,样品的峰形越细,颗粒分布越均匀[32]。因此选择破碎20 min作为SPI/KGM复合体系的微粒化条件。
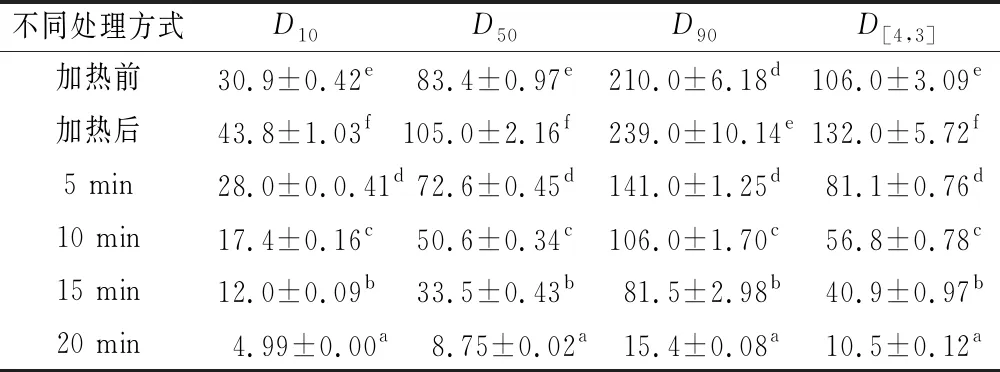
表4 脂肪模拟物制备过程粒径分布
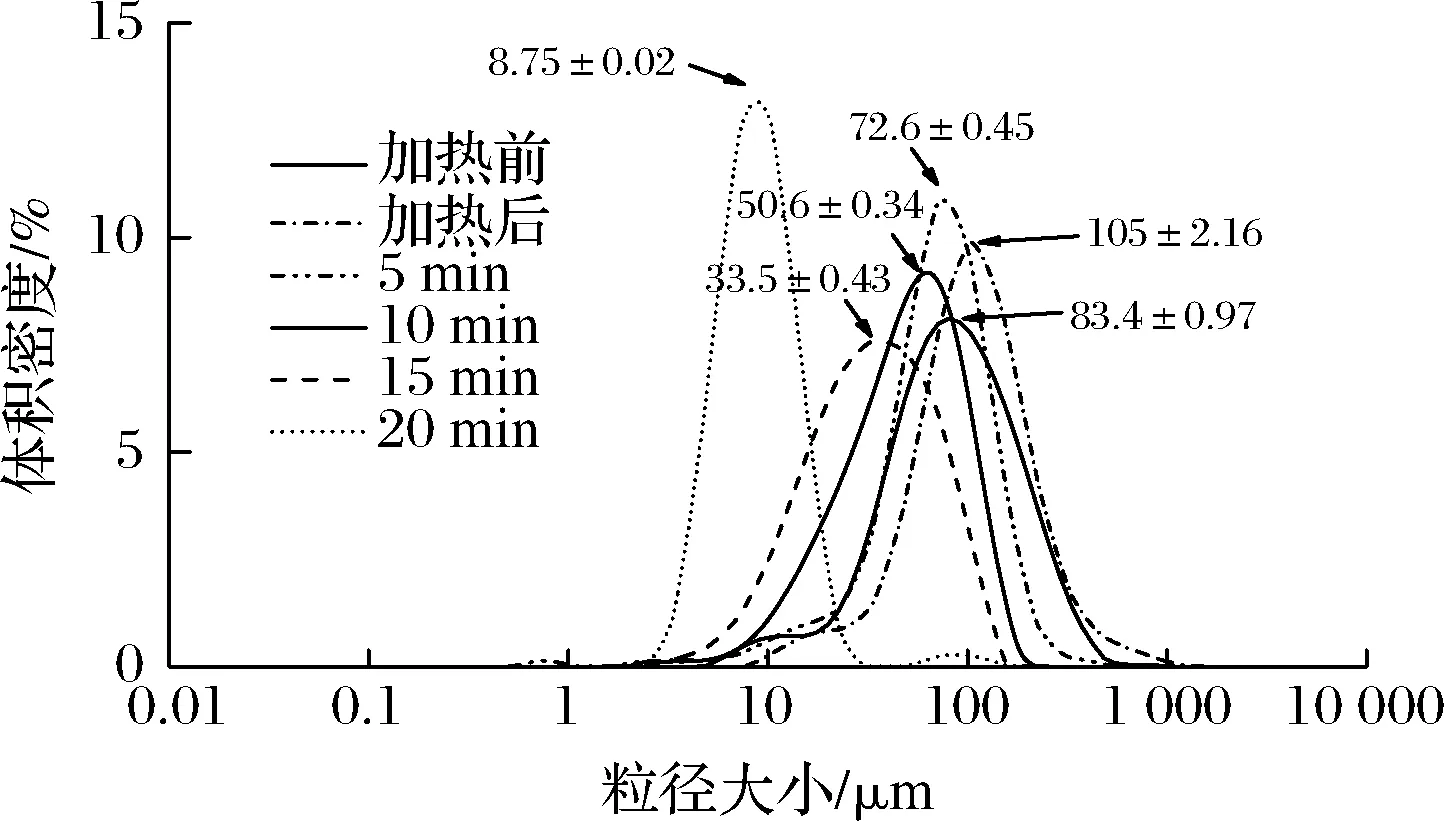
图7 不同处理方式对SPI/KGM复合体系粒径大小的影响
2.4 扫描电镜结果分析
分别对加热前、加热后、微粒化后的样品做扫描电镜观察,得到其微观结构(300×和1 000×)见图8。SPI/KGM混合物(图8-a)呈球状颗粒,混合物分散状态较好,颗粒较小且均匀。经加热变性后(图8-b),分子颗粒受热膨胀,且SPI和KGM相互运动,使得颗粒之间发生交联,形成了较为致密的海绵状三维网状结构[33],且连接处的支架结构变厚,说明体系的强度和弹性增强。样品经真空冷冻干燥后存在小孔隙,与粗大孔隙结构相比,具有小孔隙结构的体系具有更好的吸水能力和保水能力[34]。这些孔隙被SPI/KGM复合体系紧紧的包围,从而截留大量的水和营养物质的损失[35],被截留的水具有一定的流动性,感觉类似脂肪。然而蛋白质分子经湿热处理后,疏水基团和区域暴露在外,分子的亲水性降低,疏水性增强。

a-SPI/KGM复合物;b-加热后SPI/KGM复合物;c-微粒化后SPI/KGM复合物
另一方面,湿热处理过程中,物料之间发生复杂的缔合作用,形成大分子络合物,增强了脂肪模拟物的性质稳定性。但其样品表面呈粗糙,砂样质构的凝胶体,无法模拟脂肪细腻、润滑的口感。经微粒化技术处理后的SPI/KGM复合体系(图8-c)片状结构明显,表面细腻光滑,颗粒变小(<10 μm),主要是由于在高速剪切作用力下,样品颗粒变小,从而减小了食品与舌头感知的摩擦力,此时可作为脂肪模拟物。
3 结论
随着现代人对健康低脂食品的追求,脂肪模拟物得到广泛的关注。通过湿热处理制备SPI/KGM复合体系,利用单因素和响应面试验研究了制备工艺参数对复合体系膨胀率及流变特性的影响,确定最佳总固形物含量为8%、KGM添加量为0.8%、加热温度为87 ℃,在此优化条件下SPI/KGM复合体系的膨胀率可达53%,黏度可达233.3 Pa·s。试验表明,SPI/KGM复合体系的膨胀率及流变特性表现出较理想的脂肪模拟物性状,且都呈现假塑形流体特性。最后用微粒化技术处理得到的脂肪模拟物粒径<10 μm,低于人体口腔黏膜的感觉阈值,样品表面变得光滑细腻,具有良好的脂肪性状,可拓展其在低脂低热量食品的应用。复合脂肪模拟物是脂肪替代品的发展趋势,为达到更好的替代脂肪效果,在脂肪模拟物的保湿技术、香气保持、抑菌技术等方面的研究还应做进一步探索。
