掺杂、应变对析氢反应催化剂NiP2性能的影响*
2021-08-05张凤廉森王明月陈雪殷继康何磊潘华卿任俊峰陈美娜
张凤 廉森 王明月 陈雪 殷继康 何磊潘华卿 任俊峰 陈美娜
(山东师范大学物理与电子科学学院, 济南 250358)
电解水制氢可以将太阳能、风能等可持续能源中的能量转移到氢气这种高能量密度的清洁能源载体中.NiP2作为一种具有较高析氢反应(HER)催化活性的廉价催化剂而备受关注. 本文计算了NiP2(100)表面上氢的吸附能、吉布斯自由能、交换电流密度等属性, 得出其表面上P的top位点是催化活性位点的结论. 并着重分析了掺杂和应变对NiP2催化活性的影响: 1)基于常见的实验手段能产生的应变范围, 计算了1%和3%的拉伸和压缩应变的影响, 发现在NiP2上施加1%的压缩应变可以提高其HER催化性能; 2)计算了过渡金属元素及非金属元素(N, C, S)掺杂对NiP2催化性能的影响, 发现掺杂S可以显著提高P的top位点的HER催化性能, 而过渡金属元素(催化活性: Mn > Mo > W > Co > Cr > Fe > Ni)中Mn的掺杂, 可以将NiP2非活性位点的催化性能提升到跟活性位点同一个数量级, 从而间接提升NiP2的催化性能. 本工作揭示了掺杂和应变对HER催化剂性能影响的微观机理, 为设计高性能HER催化剂提供了指导.
1 引 言
化石燃料是传统的能源载体, 然而由于化石燃料的持续使用而引起的例如全球变暖和环境污染等问题, 引发了人们的持续担忧. 太阳能和风能等可再生能源的的应用是解决能源和环境问题的一种可行途径, 但是, 他们仅在阳光普照或刮风时才能供电[1]. 为了克服能源供应间歇性的问题, 发展储能技术被提上了日程[2-7]. 其中, 电化学储能技术不同于抽水蓄能等传统储能方式, 其不受地理位置和自然资源储存的限制, 是储存可再生能源的绝佳平台[5,8-14]. 电解水可将可再生能源中的能量转移到氢气和氧气中储存, 因其成本低、效率高、环境友好、可持续发展等优点被认为是最有前途和可持续发展的储能选择. 电解水包含两个半反应,一个是发生在阳极的析氧反应(oxygen evolution reaction, OER), 反应产物是氧气, 另一个是发生在阴极的析氢反应(hydrogen evolution reaction,HER), 反应产物是氢气. 本文所研究的便是发生在阴极的析氢反应.
近年来, 为了实现高效产氢, 人们对各种HER催化剂进行了系统的研究[15]. 贵金属铂被认为是最好的HER催化剂, 表现出非常小的过电位. 可惜的是, 铂的高成本和稀缺性阻碍了它在工业上的广泛使用[16]. 因此, 寻找Pt的廉价替代品作为HER的催化剂引发了人们的极大兴趣[17-19].
过渡金属化合物作为一种很有前途的非贵金属催化剂, 近年来得到了人们的广泛研究. Wang研究组[20]发现与Mo和Mo3P相比, MoP在酸性和碱性条件下都表现出较高的HER催化活性和稳定性. Aravind等[21]制备了MoP和MoP-C纳米片, 其Tafel斜率为63 mV/dec, 同样表现出了极好的HER催化活性. Wu等[22]通过简单而通用的策略合成了具有相对高比表面积(162 m2/g)的MoS2-MoP纳米片(MoS2-MoP/C), Tafel斜率为58 mV/dec, 并且在102 mV和130 mV的过电势下分别表现出的10 mA/cm2和20 mA/cm2的电流密度. Xu等[23]成功制备了Ni2P/MoP-CC双组分催化剂, 其在所有pH范围内均表现出优异的电化学性能, 在0.5 mol/L H2SO4, 1.0 mol/L PBS和1.0 mol/L KOH中分别表现出63 mV/dec,99 mV/dec和64 mV/dec的Tafel斜率和300 mV,200 mV和5 mV的起始过电势. Wang等[24]采用简单的离子交换树脂法合成了Mo2C/MoN/NG,其过电势为78.82 mV, Tafel斜率为39.3 mV/dec,而且其催化活性仅比20%Pt/C低30%左右.Harnisch等[25]发现WC在酸性条件下(300 mV下电流密度为26 mA/cm2)表现出良好的催化活性, 并比较了不同pH条件下WC的溶解速率, 发现pH越高, WC的溶解速率越快, 在一定程度上表明了WC在酸性条件下的稳定性. Ojha等[26]用石墨碳氮化物作为氮源, 制备了由5—8 nm的粒子组装而成的Mo2N纳米结构, 他们发现Mo2N纳米片显示出较高的电催化活性(在—400 mV下, 交换电流为97 mA/cm2).
近年来, NiP2由于其高HER催化活性和耐腐蚀性同样受到了广泛的关注[27]. Wang等[28]通过Ni-Mn氢氧化物作为前驱体的磷化反应成功地制备了Mn掺杂NiP2纳米片, 在0.5 mol/L H2SO4,1.0 mol/L PBS和1.0 mol/L KOH中分别表现出69 mV, 97 mV和107 mV的过电势. Tian等[29]在碳纤维纸(Al-NiP2-NSs/CFP)上合成了掺杂铝NiP2纳米片, 其表面具有超亲水性, 同时表现出了优异的析氢活性, 在0.5 mol/L H2SO4中达到10 mA/cm2只需要58 mV的低过电势. 尽管NiP2展现出卓越的HER催化性能, 其相关的理论计算却很少[28,30], 且大多停留在很基础的计算层面, 因此通过计算, 来研究如何提高NiP2的HER催化性能有很大的探索空间.
掺杂已被广泛应用于调节材料的物理和化学性质[31]. 之前的研究也表明, 掺杂是改变催化剂的催化活性的有效途径[31,32]. 然而, 掺杂对NiP2的催化活性影响尚不清楚, 且相对理论计算而言,通过实验研究来探索何种元素掺杂对NiP2的催化活性有积极的影响是一项耗时耗力且成本较高的工作.
值得注意的是, 近年来, 人们发现通过调节催化剂原子的膨胀或压缩排列, 从而调整其表面电子结构[33,34], 是提高催化剂材料HER催化活性的一种很有前途的方法[35-37]. 据我们所知, 当前关于应变对HER催化剂材料性能影响的理论研究非常稀少, 尽管实验上可以通过很多措施达到在HER催化剂材料上施加应变的效果[38-40], 但其具体应变调控策略非常模糊, 尚没有找出明确的规律.
因此, 本文着重研究了掺杂和应变对NiP2HER催化性能的影响机理, 从一个新的角度给实验工作者提高HER催化剂的性能提供了思路, 为设计出高性能HER催化剂提供了帮助.
2 计算方法
本文基于第一性原理计算方法, 使用VASP(Viennaab initiosimulation package)软件包进行模拟[41]. 计算中选择了PAW(projection enhanced wave)方法描述离子与电子之间的相互作用[41], 根据PBE (Perdew-Burke Ernzerhof)泛函的广义梯度近似(GGA)选择交换相关势[42],文中涉及元素的有效价电子分别为: Ni(3d84s2),Co(3d74s2), Fe(3d64s2), Mn(3d54s2), W(5d46s2),Mo(4d55s1), Cu(3d104s1), Cr(3d54s1), N(2s22p3),C(2s22p2), P(3s23p3), S(3s23p4), H(1s1). 平面波截断能设置为450 eV, 能量自洽收敛条件为10—5eV.当作用在每个原子上的Hellmann-Feynman力小于0.03 eV/Å (1 Å = 0.1 nm)时, 认为达到力的收敛条件. 高斯展宽设置为0.05 eV. 采用以Г点为中心的3 × 3 × 1 的k点网格进行布里渊区积分. 热力学函数值的分析通过能源材料高通量计算平台来完成[43].
3 结果与讨论
3.1 晶格结构
基于已有实验结果[28], 选择NiP2(JCPDS卡片编号为JCPDS No. 21-0590)的(100)表面进行了研究. NiP2的原胞(a=b=c= 5.453 Å,α=β=γ= 90°, 空间群:pa3)由12个原子组成,其中镍原子4个, 磷原子8个. 在计算材料的表面性质时, 建立2 × 2 × 2包含96个原子的超胞,NiP2(100)表面模型(见图1)是由4层原子组成的slab模型, 垂直于(100)表面的真空层厚度为1.5 nm. NiP2(100)表面上有5个H吸附位点, 分别用位点1 (P的top位点), 位点2 (Ni的top位点), 位点3 (第一层中更加接近表面的P-P之间的bridge位点), 位点4 (第一层中稍微远离表面的P-P之间的bridge位点)和位点5 (Ni-Ni之间的bridge位点)来表示.
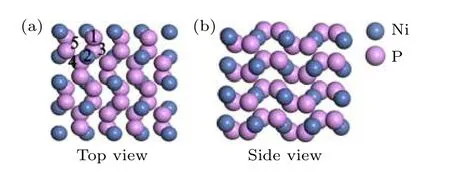
图1 (a) NiP2 (100)表面的俯视图和NiP2 (100)表面的不同吸附位点; (b) NiP2 (100)表面的侧视图Fig. 1. (a) Top view of NiP2 (100) surface and different adsorption sites on NiP2 (100) surface; (b) side view of NiP2(100) surface.
3.2 NiP2(100)表面的吸附能
氢原子吸附在催化剂表面的吸附能(adsorption energy, ΔE)可由以下等式计算[44,45]:
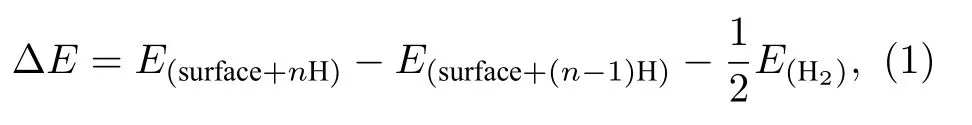
式中,E(surface+nH)表示表面吸附n个H的NiP2的体系总能量.E(surface+(n-1)H)是表面吸附n—1个H的NiP2的体系总能量,E(H2)是气相H2的总能量.吸附能是描述H在催化剂表面吸附能力的一个重要描述符[19]. 当ΔE小于0时, 表明H能在催化剂表面自发吸附, 此时ΔE值越小, 催化剂表面对H的吸附能力越强; 当ΔE大于0时, 说明H不能在催化剂表面自发吸附, 此时ΔE值越大, 催化剂表面对H的吸附能力越弱.
在ΔE的计算中, 首先将H分别放在NiP2(100)表面的不同吸附位点上, 得到催化剂表面(100)吸附H后的且优化后的体系总能量E(surface+nH).同时, 对未吸附H的干净的(100)表面进行了优化, 得到了干净表面优化后的体系总能量E(surface+nH)(n=0).E(H2)是气相H2优化后的体系总能量. 将上述3个结果代入(1)式, 可得到(100)表面上的不同吸附位点的ΔE.
通过对H吸附在不同位点的吸附结构进行优化, 发现优化前吸附在位点3的H在进行结构优化后会移动到位点1上, 表明H无法稳定吸附在(100)表面的位点3上, 而吸附在位点4和位点5上的H在进行结构优化后同样产生了吸附位置的偏移, 表明H同样无法稳定吸附在(100)表面的位点4和位点5上, 但为了方便进行相关位点的表示, 在之后的计算中, 仍用位点4 (第一层中稍微远离表面的P-P之间的bridge位点)和位点5(Ni-Ni之间的bridge位点)来表示偏移后的H吸附位置.
根据表1可以看出, 当H吸附在位点1的时候, 吸附能小于0 (ΔE= —0.209 eV), 表明H与P的top位点的结合能力较强, 这是由于非金属原子P的非金属性导致P原子对电子有很强的吸引力, 使得电子聚集在其周围, 进而吸引电解质溶液中带正电荷的质子H靠近P原子, 使H牢固地吸附在P的top位点上. 而当H吸附在位点2时, 吸附能大于0 (ΔE= 0.674 eV), 表明H与Ni的top位点的结合能力较弱, 这是因为过渡金属原子Ni具有一定的金属性质, 表现为正电性, 而电解质溶液中的质子H也带有正电荷, 因此, 过渡金属原子Ni与H相互排斥, 表现为对H较弱的吸附能力. 因此, 位点1 (P的top位点)是NiP2(100)表面可以稳定吸附H的位点.
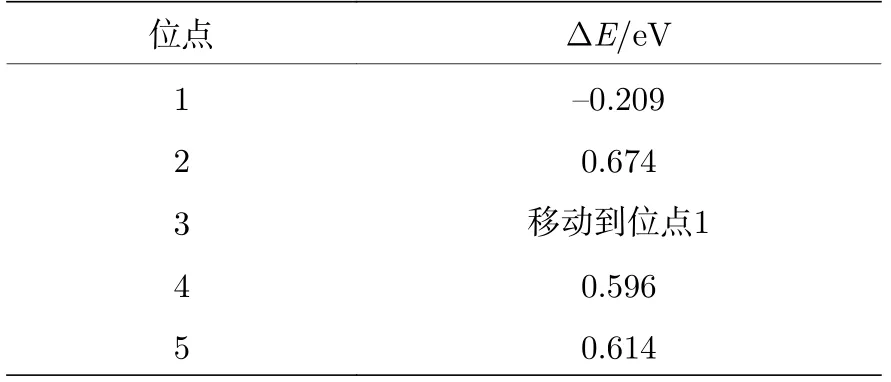
表1 NiP2 (100)表面的不同位点的吸附能ΔETable 1. Adsorption energy (ΔE ) of different sites of NiP2 (100) surface.
3.3 NiP2 (100)表面的吉布斯自由能
吉布斯自由能(ΔG)是一个描述催化剂材料HER催化活性的一个重要描述符[46,47]. ΔG可以通过以下公式进行计算[19,48]:
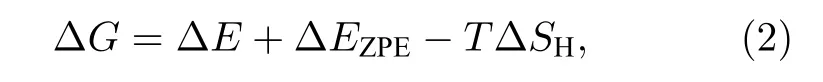
式中ΔE为H吸附在催化剂表面的吸附能, ΔEZPE为H吸附状态与气相状态下零点振动能之差, 其由下式表示:
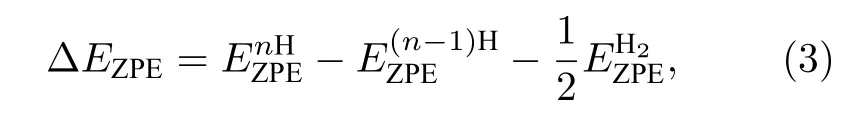
式中,表示n个H吸附在NiP2(100)表面的零点能,表示n—1个H吸附在NiP2(100)表面上的零点能,表示气相H2的零点能. 每个零点能可以通过方程获得, 其中h表示普朗克常数,νi表示振动频率, ΔSH表示吸附态氢原子与气相中氢原子之间的熵差, 其可以通过此公式获得:其中表示在标准状况下气相下H2的熵.
当ΔG远小于0时, 表明催化剂表面吸附的H与催化剂表面的结合较强, 进而使H2的生成和脱附变得困难; 当ΔG远大于0时, 表明催化剂表面吸附的H与催化剂表面结合较弱, 从而难以形成H2; 当ΔG接近0时, 表明H的吸附和H2的脱附达到平衡, 进而在催化剂表面发生析氢反应. 因此, 本工作计算了NiP2(100)表面的不同吸附位点的吉布斯自由能(见图2), 进而判断哪个位点是NiP2(100)表面最具有HER催化活性的位点.
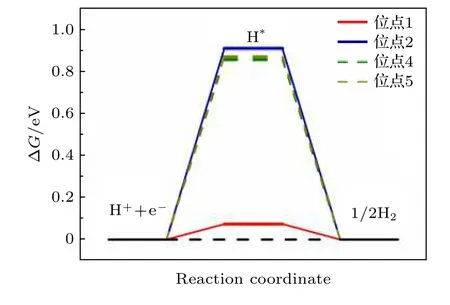
图2 NiP2 (100)表面不同吸附位点的ΔG, 虚线表示H吸附位点的偏移Fig. 2. ΔG on different adsorption sites on NiP2 (100) surface, the dashed line indicates the shift of H adsorption site.
从图2可以看出, 当H吸附在位点1(P的top位点)时, ΔG接近于0 (ΔG= 0.074 eV), 表现出极好的HER催化活性, 而当H吸附在位点2(Ni的top位点)的时候, ΔG远离0 (ΔG= 0.906 eV),表现出较弱的HER催化活性. 而图2中的虚线部分表示H偏离结构优化前的吸附位点4(第一层中稍微远离表面的P-P之间的bridge位点)和位点5(Ni-Ni之间的bridge位点)后, 所展现出的ΔG,同样表现出很弱的HER催化活性. 可认为不同的吸附位点表现出如此迥异的HER催化性能是由以下原因导致的: 当H吸附在位点1 (P的top位点)的时候, 吸附能小于0 (ΔE= —0.209 eV), 表明H可以较为容易地吸附在NiP2(100)表面上, 同时吸附能并没有非常小, 同样可以实现较为容易地H2的脱附, 因此表现出较好的HER催化活性.而当H吸附在其余位点的时候, 吸附能远大于0(位点2: ΔE= 0.674 eV; 位点4: ΔE= 0.596 eV;位点5: ΔE= 0.614 eV), 表明H吸附在这些位点上较为困难, 不容易形成H2, 更不容易发生析氢反应, 进而表现出较弱的HER催化活性. 因此, 从H的吸附能和吉布斯自由能的角度看, P的top位点是NiP2中参与HER催化反应的主要催化位点.
3.4 交换电流密度
交换电流密度(exchange current density,j0)是表征催化剂催化活性的重要指标, 对整个电化学反应速率有着深远的影响. 交换电流密度可以通过以下公式进行计算[49,50]:
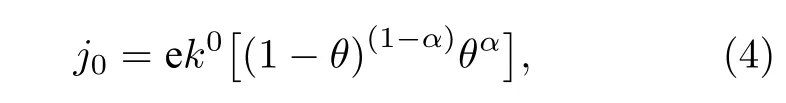
其中j0为单个位点的理论交换电流密度,e为单个电子所带电荷,k0为标准速率常数, 为200 s—1·site—1,α是传递系数,θ是与吸附热相关的一个量. 高性能的催化剂应该具有中等的吸附能力, 如果吸附太弱, 反应物不会吸附在表面, 而如果吸附太强, 则脱附困难. 在吸附能与催化速率的关系图上, 往往都是呈现出一种先上升后下降的火山形状的曲线,因而形象地称之为火山型曲线. 通常, 合适的吸附能应位于火山型曲线的顶点附近. 为了进一步研究不同位点的催化性能, 比较了不同位点的交换电流密度相对于火山型曲线顶点的位置. 当电荷转移系数α= 0.45时, NiP2(100)表面ΔG与j0的火山型曲线见图3.
从图3可以看出, 当H吸附在位点1 (P的top位点)的时候, 理论计算出的交换电流密度最大,此时的交换电流密度接近于火山型曲线的顶点, 表现出极好的HER催化活性. 而当H吸附在其余吸附位点(位点2 (Ni的top位点), 位点4 (第一层中稍微远离表面的P-P之间的bridge位点)和位点5 (Ni-Ni之间的bridge位点))的时候, 交换电流密度集中出现在火山型曲线的右下侧, 距离具有最好HER催化活性的火山图顶点有很大一部分距离, 表现出极弱的HER催化活性. 这一结论跟我们之前从吸附能和吉布斯自由能分析出P的top位点具有最佳的HER催化反应活性是一致的.
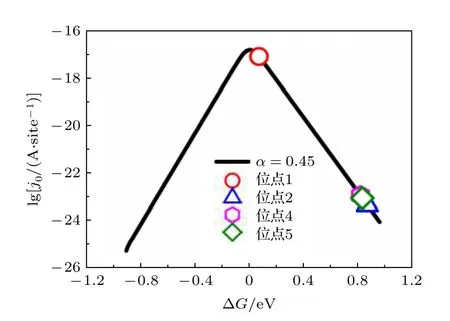
图3 当电荷转移系数α = 0.45(黑色曲线)时, NiP2 (100)表面ΔG与 j0 的火山型曲线, 空心符号表示通过吉布斯自由能获得的单个位点的理论交换电流密度Fig. 3. Volcano plot between j0 and ΔG with charge-transfer coefficient α = 0.45 (black solid line) of NiP2 (100) surface. The hollow symbols represent the theoretical exchange current density of a single site obtained by the value of Gibbs free energy.
3.5 掺杂对NiP2 (100)表面HER催化活性的影响
本文计算了过渡金属元素(Co, Fe, Mn, Mo, Cu,W, Cr)掺杂和非金属掺杂(N, C, S)对NiP2(100)表面HER催化性能的影响. 其中, 图4(a)表示对NiP2进行过渡金属掺杂和非金属掺杂后, 位点1(P的top位点)的ΔG变化, 图4(b)表示对NiP2进行过渡金属掺杂前后, H分别吸附在位点2(Ni的top位点)和掺杂过渡金属原子上的ΔG, 图4(c)表示对NiP2进行非金属掺杂前后, H分别吸附在位点1(P的top位点)和掺杂非金属原子上的ΔG.
从图4(a)可以看出, 相比于掺杂前的位点1(P的top位点)的ΔG, 仅掺杂非金属元素S后,ΔG更加接近于0 (ΔG= —0.007 eV), 表现出增强的HER催化性能, 而掺杂过渡金属元素Mn, Mo,W, Co, Cr, Fe, Cu及非金属元素N, C却对此位点的催化性能没有帮助.
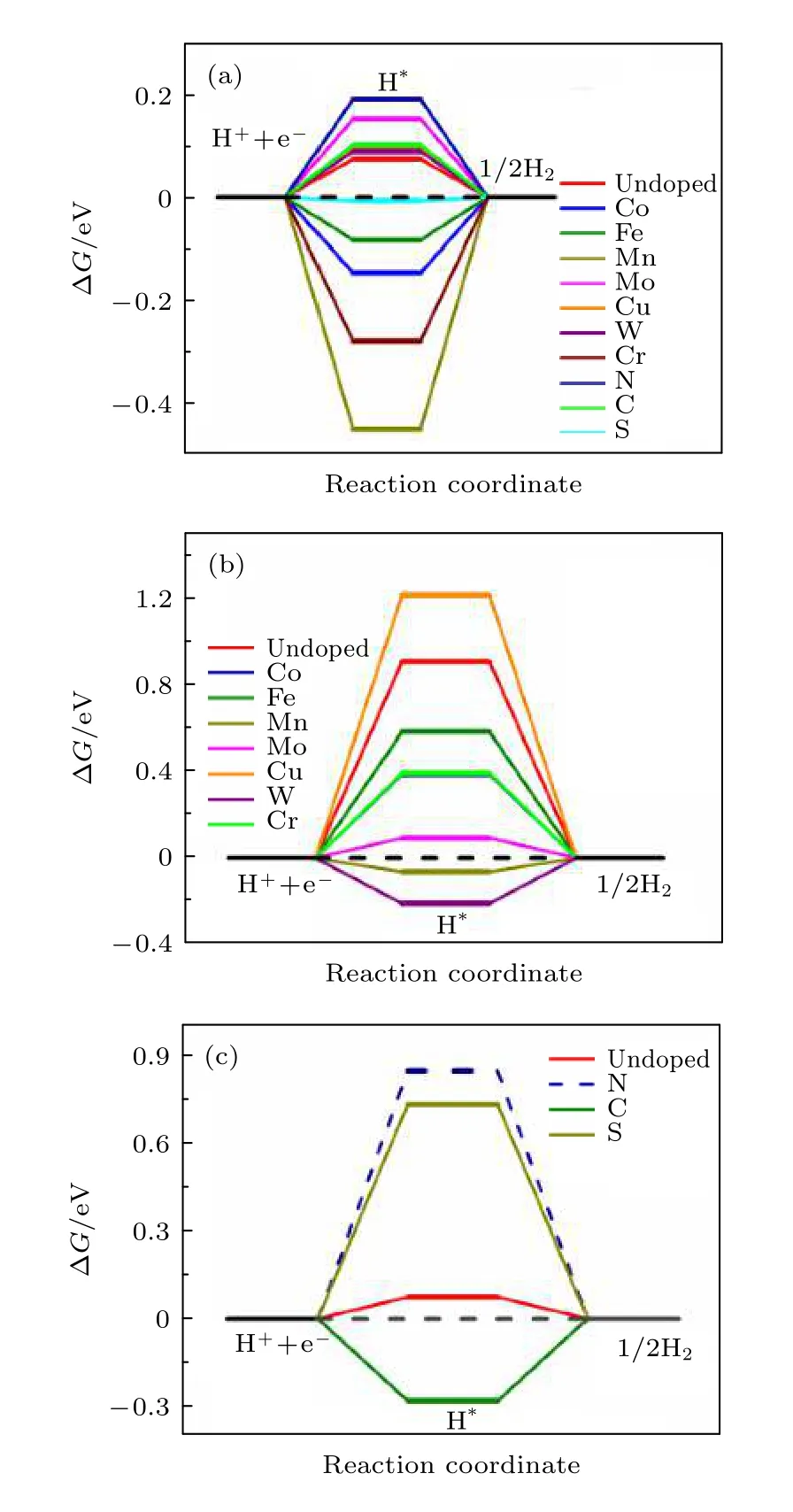
图4 (a) 过渡金属和非金属元素掺杂前后H吸附在NiP2(100)表面位点1(P的top位点)上和掺杂原子上的ΔG;(b) 过渡金属掺杂前后H吸附在NiP2(100)表面位点2(Ni的top位点)上和掺杂的过渡金属原子上的ΔG; (c)掺杂非金属原子前后H分别吸附在NiP2(100)表面位点1(P的top位点)和掺杂的非金属原子上的ΔG; 虚线表示H吸附位点有所偏移Fig. 4. (a) ΔG of H adsorbed on site 1 (top site of P atom)of NiP2 (100) surface before and after doping, (b) ΔG of H adsorbed on site 2 (top site of Ni atom) and top site of doped transition metal atom before and after doping transition metal atom respectively, (c) ΔG of H adsorbed on site 1 (top site of P atom) and top site of doped non-metallic atom on NiP2 (100) surface before and after doping nonmetallic atom respectively. The dashed line indicates the shift of H adsorption site.
有意思的是当H吸附在掺杂的过渡金属原子上时, 除了掺杂Cu原子后的NiP2(100)表面的催化活性没有明显改变, 掺杂过渡金属原子Co, Fe,Mn, W, Mo和Cr后的NiP2(100)表面的催化活性相比于未掺杂前的NiP2(100)表面的位点2(Ni的top位点)的催化活性有明显提升, 尤其是掺杂Mn原子后, ΔG由未掺杂前的0.906 eV变为—0.065 eV, Mn原子的掺杂将NiP2非活性位点的催化性能提升到跟活性位点(ΔG= 0.074 eV)同一个数量级, 从而间接提升NiP2的催化性能.
而从图4(c)可以看出, 当H吸附在掺杂的非金属原子上时, 催化活性的改变却不尽如人意, 相比于未掺杂前H直接吸附在非金属原子P上(位点1)表现出的很好的催化活性(ΔG= 0.074 eV),H吸附在掺杂的非金属原子C原子和S原子上时,ΔG更加远离0, 表示HER催化性能的降低, 同时可发现H无法稳定吸附在掺杂的N原子上, 因为对H吸附在掺杂N原子的结构模型进行优化后,原本吸附在N原子上的H移动到表面其余位置.因此, 掺杂非金属元素S可以显著提高催化活性位点(P的top位点)的析氢反应催化性能, 而过渡金属元素(催化活性: Mn > Mo > W > Co > Cr >Fe > Ni)中Mn的掺杂可以间接提升NiP2的催化性能.
3.6 应变对NiP2 (100)表面HER催化活性的影响
为了研究应变对NiP2(100)表面HER催化活性的影响, 基于实验上实际能达到的施加在催化剂材料上的应变程度[51-53], 分别计算了1%和3%的拉伸和压缩应变下NiP2(100)表面催化活性位点1 (P的top位点)的ΔG, 目的是探究应变对HER催化性能的影响, 预测可以提高NiP2(100)表面的催化活性的最佳应变. 通过增加或者减少NiP2(100)表面晶格常数的数值, 来实现在催化剂上施加拉伸或压缩应变的目的, 表面不同应变的NiP2(100)的ΔG如图5所示.
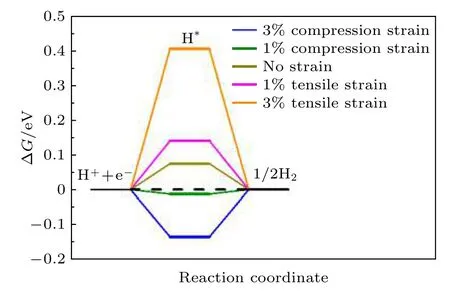
图5 NiP2 (100)表面不同应变的ΔGFig. 5. ΔG with different strain on NiP2 (100) surface.
从图5可以看出, 对NiP2(100)表面施加1%的压缩应变时, NiP2(100)表面的ΔG由0.074 eV变为了—0.013 eV, 即进行1%压缩应变后的ΔG相较于没有施加应力之前更加接近0, 表现出更好的HER催化性能. 而对NiP2(100)表面施加3%的压缩应变或1%, 3%的拉伸应变时, 其相应的ΔG反而相较于没有施加应变之前更加远离0, 这说明施加其他程度的应变并不能增加NiP2(100)表面的催化活性. 因此, 想要提高HER催化剂的催化活性, 选择合适的应变非常重要. 实际上, 有许多实验方法可以实现在催化剂材料上施加应变的效果, 包括人为制造晶格失配[33,54]、原子层面上控制材料厚度[54]、阳离子交换法[34]和施加外力[38,39,55]等. 这些实验方法都为理论预测结果的实现提供了可靠的实验实现途径.
4 结 论
本文利用第一性原理方法, 计算了NiP2最稳定(100)表面不同位点上氢的吸附能、吉布斯自由能、交换电流密度等属性, 并在此基础上着重研究了掺杂和应变对NiP2HER催化性能的影响. 通过计算, 发现当H吸附在NiP2(100)表面上P的top位点上时, 其交换电流密度值最接近交换电流密度火山图的顶点, 因而P的top位点是NiP2的HER催化活性位点. 通过研究过渡金属(Co, Fe, Mn,W, Mo, Cu, Cr)以及非金属(N, C, S)掺杂对NiP2的催化性能的影响, 发现掺杂非金属元素S可以显著提高P的top位点的HER催化性能, 而掺杂过渡金属元素Mn, Mo, W, Co, Cr, Fe, Cu及非金属元素N, C却对此位点的催化性能没有帮助. 而过渡金属元素(催化活性: Mn > Mo > W > Co >Cr > Fe > Ni)中Mn的掺杂, 可以将NiP2非活性位点的催化性能提升到跟活性位点同一个数量级, 从而间接提升NiP2的催化性能. 基于常见的实验手段能产生的应变范围, 本工作还计算了1%和3%的拉伸和压缩应变的影响, 发现在NiP2上施加1%的压缩应变可以提高其HER催化性能.
本文揭示了掺杂、应变提高NiP2HER催化活性的微观机理, 从新的角度为设计和研发催化性能更好的HER催化剂提供了理论指导.
