基于亲水作用色谱-串联质谱的非衍生化法测定绿茶中草甘膦、草铵膦及其代谢物残留
2021-08-03邱世婷贺光云覃蜀迪
邱世婷 侯 雪 韩 梅 李 莹 贺光云 覃蜀迪 沈 璐
(四川省农业科学院农业质量标准与检测技术研究所,农业农村部农产品质量安全风险评估实验室(成都),成都 610066)
在茶园管理中,除草是一项贯穿整个生产过程的农事活动,草甘膦(glyphosate,GLY)和草铵膦(glufosinate,GLU)是我国登记允许在茶园使用的除草剂[1],但近年来,滥用草甘膦等引发的茶叶质量安全事故引起消费者越来越多的关注。草甘膦具有与有机磷化合物相似的作用毒理,主要通过抑制胆碱脂酶的活性而导致神经系统机能失调[2]。草铵膦的主要代谢产物为3-(甲基膦基)丙酸(3-methylphosphinico propionic acid,MPP)和N-乙酰草铵膦(N-acetyl glufosinate sodium,NAG)。草铵膦对人体的生殖系统会产生毒性,其代谢产物也具有相似毒性[3],存在一定风险,因此同时检测草铵膦及其代谢物十分必要。许多国家和组织已制定草铵膦及其代谢物的最大残留限量,国际食品法典委员会(CAC)、欧盟、日本和韩国规定草铵膦的残留定义为草铵膦、MPP和NAG之和[4~6],我国GB 2763-2019《食品安全国家标准 食品中农药最大残留限量》规定草铵膦的残留仅为草铵膦。欧盟、美国、日本和中国对茶叶中草甘膦的最大残留限量分别为2.0、1.0、1.0和1.0 mg/kg,欧盟、日本和中国对茶叶中草铵膦的最大残留限量分别为0.1、0.3和0.5 mg/kg。
草甘膦、草铵膦及其代谢物结构类似,含有膦酸基、羟基、氨基,为强极性的两性化合物,不溶于大部分有机试剂,无发色团或荧光基团,难气化,在反相液相柱上无保留,采用常规手段对其进行定性定量极困难[8~10]。目前国内外测定绿茶中草甘膦、草铵膦及其代谢物通常采用以9-芴基甲基氯仿(9-fluorenylmethyl chlorocarbonate,FMOCCl)为衍生化试剂,通过衍生改善其色谱保留行为,从而可被仪器检测的方法,但衍生化法操作复杂、耗时长,同时衍生副产物易污染仪器,干扰测定,重现性差[11~13]。近年来,不经衍生直接采用液质联用测定食品中草甘膦和草铵膦的方法也有报道[14~15],如YASUSHI等[16]采用InertSep SAX柱净化,对啤酒、大麦茶中的草甘膦和草铵膦及其代谢物进行了测定;BOTERO-COY等[14]采用HLB柱对大豆、玉米样品中的草甘膦和氨甲基膦酸进行了测定;孙文闪等[17]采用MWCNTs为净化剂,对茶叶中草甘膦、氨甲基膦酸、草铵膦进行了测定。但大部分研究集中在草甘膦、草铵膦原药,少见涉及代谢产物NAG和MPP检测的报道,也未见采用非衍生化法同时检测绿茶中草甘膦、草铵膦及其代谢物残留的报道。
基于草甘膦和草铵膦的强极性和阴离子特性,本研究采用亲水作用色谱-串联质谱,结合HLB柱净化,建立了一种无需衍生,净化步骤简单,能同时测定绿茶中草甘膦、草铵膦及其代谢物MPP和NAG残留量的方法,以期为绿茶中的草甘膦、草铵膦及其代谢物的快速测定提供参考。
一、材料与方法
(一)仪器与试剂LC-30A超高效液相色谱8060三重四极杆质谱仪(日本岛津公司);Multi Reax多管涡旋振荡器(Heidolph公司);AUY220电子天平(日本岛津公司);Neofuge 18R台式高速冷冻离心机(上海力康仪器有限公司);MilliQ超纯水器(美国Millipore公司)。
GLY、GLU、MPP、NAG标 准 品(纯 度>98%,天津阿尔塔科技有限公司);PRiME HLB小柱(200 mg/6 mL,美国沃特世公司);实验用水为超纯水;乙腈、甲酸(色谱纯,美国Fisher Scientific公司)。
(二)实验方法
1.标准溶液的配制。准确称取10 mg的各农药标准品,分别用超纯水溶解并配制成质量浓度为100 mg/L的单个标准储备液,于4℃冰箱保存,有效期3个月。分别吸取0.2 mL单个标准储备液于10 mL容量瓶中,用水定容至10 mL,配制成质量浓度为2 mg/L的混合标准储备液,放置于4℃冰箱,有效期1个月。使用时,根据需要用超纯水或空白基质配制成适合浓度的上机工作溶液。
2.样品前处理。称取粉碎的绿茶样品1.0 g,置于50 mL离心管中,加入10 mL超纯水提取,涡旋振荡20 min,8 000 r/min离心5 min,取2 mL通过PRiME HLB小柱,收集流出液,过0.22μm滤膜,供超高效液相色谱串联质谱(ultra performance liquid chromatography tandem mass spectrometry,UPLC-MS/MS)测定。
3.液相和质谱条件。(1)液相色谱条件:色谱柱为Waters阴离子极性农药(anionic polar pesticide)色谱柱(100 mm×2.1 mm,5μm);柱温为50℃;流速为0.5 mL/min;进样量为10μL;流动相A为0.9%甲酸水溶液,流动相B为0.9%甲酸-乙腈。梯度洗脱程序:0~4 min,0%B~15%B;4~7 min,15%B;7~8 min,15%~90%B;8~12 min,90%B。(2)质谱条件:质谱离子源为电喷雾电离(electron spray ionization,ESI)源;接口电压为4 000 V;多反应监测(multiple reaction monitoring,MRM)模式;雾化气流速为3 L/min;干燥气流速为10 L/min;加热气流速为10 L/min;接口温度为300℃;脱溶剂管温度为250℃;加热模块温度为400℃。其他质谱参数见表1。

表1 4种化合物的质谱参数
二、结果与分析
(一)质谱条件的优化单独配制质量浓度为1 mg/L的4种化合物的标准溶液,进行Q1全扫描,经扫描发现,目标化合物在负离子模式下响应强度高、稳定性好,在负离子模式下确定4种化合物的母离子峰均为[M-H]-峰,将其作为母离子,利用仪器自带自动优化程序,对子离子、碰撞电压、Q1电压、Q3电压进行优化,获得二级质谱优化参数,最终形成的MRM扫描参数见表1。
(二)色谱条件的优化草甘膦、草铵膦及其2种代谢物在传统的反相柱中基本无保留,根据其高亲水性和强极性特点,本实验研究选择亲水作用色谱柱进行色谱分离,共评估了4种色谱柱:Asahipak-NH2P-50柱、Dikma polyamino氨基柱、Waters DEA柱、Waters anionic polar pesticide柱。4种色谱柱液相分离条件见表2,0.05 mg/L草甘膦、草铵膦及其代谢物在不同色谱柱上色谱分离图见图1。可见使用Asahipak-NH2P-50柱、Dikma polyamino氨基柱时,4种化合物分离效果差,基本在同一时间出峰,不能达到基线分离,且基质干扰严重,灵敏度低。使用Waters DEA柱时,草甘膦色谱峰拖尾。使用Waters anionic polar pesticide柱时,4种化合物分离效果好,峰型尖锐对称且灵敏度高。Waters anionic polar pesticide柱固定相由具有三键键合二乙胺(diethylamine,DEA)键合相的亚乙基桥杂化(bridge ethylidene hybrid,BEH)颗粒组成。这种键合相兼具亲水性表面和阴离子交换特性,适合用于保留和分离极性阴离子化合物。4种化合物均有强阴离子膦酸盐基团,在弱酸性溶液中为强电离状态带负电,通过阴离子交换保留在柱上,用0.9%甲酸的酸性流动相洗脱阴离子,4种目标化合物分离效果和峰形均较好,加入盐(如甲酸铵)后反而对峰分离效率和峰形状产生了不利影响,并导致分析物的灵敏度降低,故本研究选取Waters anionic polar pesticide柱作为分析色谱柱,含0.9%甲酸的水和乙腈为流动相。
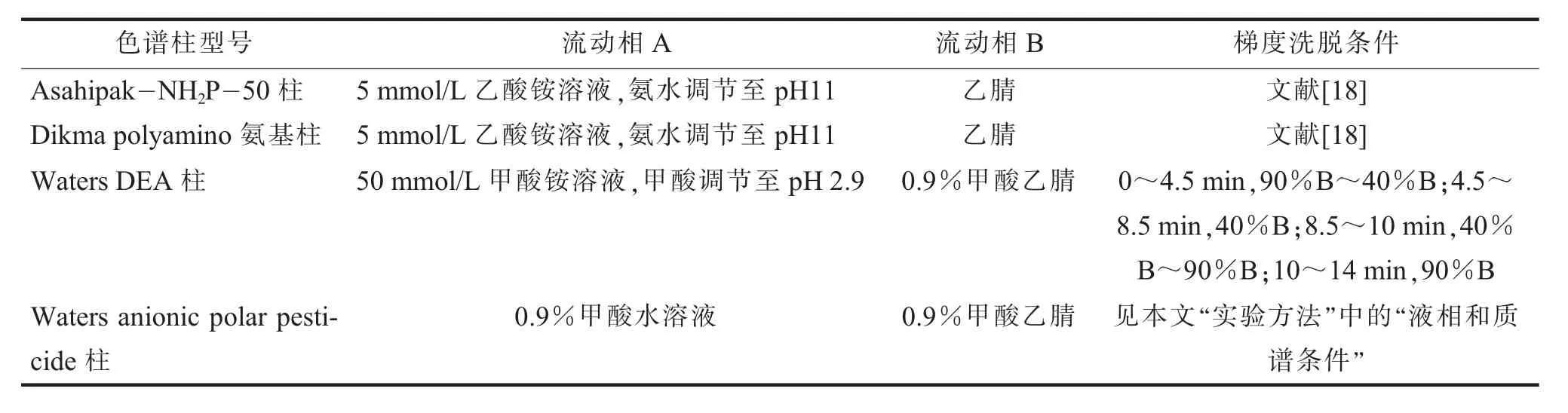
表2 4种色谱柱液相分离条件
(三)前处理条件的优化考虑绿茶基质复杂,含有大量的氨基酸、茶多酚、咖啡因、色素等干扰物质,本研究参照文献[17]前处理方法比较了50 mg MWCNTs、50 mg GCB作为吸附剂,和本文PRiME HLB小柱方法,得出空白基质净化后的负离子模式下全扫描图(见图2),结果可见HLB小柱净化基质干扰较少。PRiME HLB小柱填料为亲水亲脂反相吸附剂,能较好吸附绿茶中茶多酚、咖啡因、氨基酸等杂质[19]。采取过滤式净化模式,SPE柱无需活化、淋洗和洗脱步骤,绿茶样品经纯水提取后,取上清液直接过小柱,收集洗脱液即可上机测定,4种目标化合物回收率为78.8%~101.5%。该法操作简单,且能有效去除绿茶中的基质干扰,无需用到有机试剂,绿色环保,因此本研究选择PRiME HLB小柱作为净化方式。

图2 不同前处理方式净化后质谱全扫描色谱图
(四)方法分析性能
1.线性关系、检出限和定量限。采用空白绿茶基质配制一系列标准上机溶液,以目标化合物峰面积(Y)为纵坐标,相应的质量浓度(X,μg/L)为横坐标计算线性关系,以3倍信噪比(S/N)计算检出限(limit of detection,LOD),以10倍信噪比(S/N)计算定量限(limit of quantitation,LOQ),结果见表3。在各自的相应范围内,4种化合物线性关系良好,相关系数均>0.999,方法检出限为0.01~0.05 mg/kg;定量限为0.05~0.10 mg/kg,方法灵敏度高。

表3 4种化合物在绿茶中的线性关系、检出限、定量限及基质效应
2.回收率与精密度。分别在LOQ、2LOQ、10LOQ 3个浓度水平下对空白绿茶进行添加回收实验,每个浓度水平平行6次。4种化合物的加标回收率和相对标准偏差结果见表4。由表4可见,4种化合物回收率在78.8%~101.5%,相对标准偏差(RSD)为4.3%~11.1%,该方法的回收率和相对标准偏差满足农药残留检测中对准确度和精密度的要求。草甘膦、草铵膦及其代谢物在绿茶样品中加标回收(添加量0.2 mg/kg)及绿茶空白样品图谱见图3。

图3 草甘膦、草铵膦及其代谢物在绿茶样品中加标回收(添加量0.2 mg/kg)及绿茶空白样品图谱

表4 4种化合物的加标回收率和相对标准偏差(n=6)
3.基质效应。取绿茶空白基质溶液和试剂配制4种化合物的标准溶液,分别测定溶剂中目标农药的响应值(A)与空白基质中添加相同浓度的目标农药的响应值(B),基质效应(matrix effect,ME)=(B/A-1)×100%,ME值为负数,说明存在基质抑制效应;ME值为正,说明存在基质增强效应。实验结果表明,4种化合物在绿茶均存在不同程度基质抑制效应,其中草铵膦为强抑制效应(见表3),因此本研究采用基质匹配标准溶液外标法定量。
4.与其他方法的比较。本研究将本方法与文献报道[1,18,20~22]的 前处理 方 法在 消 耗时间、 有机 试剂消耗体积、定量限等方面进行了比较(见表5)。可知本方法在时间成本、有机试剂消耗量和定量限等方面有明显的优势,且操作简单,可满足实际样品的检测要求。
5.实际样品的检测。应用该方法对四川省市场上的67个绿茶样品进行了检测,结果显示,67批次绿茶中草铵膦及其代谢物均未检出,但40个样品检出草甘膦(>LOD),检出率超过50%,定量检出(>LOQ)范围为0.050 0~0.585 0 mg/kg,均未超过GB 2763-2019《食品安全国家标准 食品中农药最大残留限量》中的规定限量值,说明茶园中使用草甘膦情况较为普遍,应当加以管控,避免绿茶中草甘膦残留对人体造成危害。
三、结 论
本研究建立了PRiME HLB小柱净化,结合亲水作用色谱-串联质谱同时测定绿茶中草甘膦、草铵膦、3-(甲基膦基)丙酸,N-乙酰草铵膦残留的方法。本方法前处理简单,无需衍生,对环境友好,干扰少,重现性好,可作为大批量绿茶中草甘膦、草铵膦及其代谢物的分析检测方法。
