Ⅳ型Waardenburg综合征合并孤立肾1例并文献复习
2021-07-28于新平周敬淳柯朝阳
鄢 敏 张 搏 于新平 周 舟 周敬淳 柯朝阳,* 刘 明
1.暨南大学第二临床医学院(深圳市人民医院)耳鼻喉科 (广东 深圳 518020)
2.深圳市人民医院龙华分院急诊科 (广东 深圳 518109)
Waardenburg综合征(Waardenburg syndrome,WS)又称听力-色素综合征,在1951年首次被荷兰眼科学家提出,是一种较少见的常染色体遗传病,多为显性遗传[1]。目前认为WS可能的病因是神经嵴细胞发育缺陷及障碍。WS的主要临床表现有感音神经性听力异常和虹膜、头发、皮肤色素分布异常。
1 资料与方法
患儿,男,3岁1月,病例特点:产检时发现患儿右肾缺如,左侧孤立肾;出生后呕吐,入住新生儿科,腹部平片提示肠梗阻,后诊断先天性巨结肠,3+月龄时手术切除部分结肠;出生后听力筛查未能通过;出生后眼科诊断双眼异色虹膜。父母及姐姐听力正常,无虹膜异色征、前额白发等,3代内直系家属均无类似病史。
1.1 专科检查情况患儿出生后行畸变产物耳声发射检查及自动听性脑干反应检查,双耳转诊;满月后复查仍双耳转诊。3月龄对听力进行诊断评估:听性脑干反应检查提示双耳反应阈大于95dBnHL,多频稳态诱发电位检查双耳异常,1000Hz鼓室图双耳A型图,镫骨肌声反射未引出。12+月龄内耳CT示:双侧内耳发育畸形,双侧半规管、前庭池及双侧耳蜗不足2.5圈;内耳MRI示:双侧内耳发育畸形,双侧上半规管短宽、后半规管细小,双侧前庭池形态失常,双侧耳蜗周数未达2.5圈,蜗窗明显狭小近似封闭。
1.2 专科处理3+月龄完成诊断型听力评估后开始康复治疗,6+月龄佩戴助听器,13+月龄行右耳人工耳蜗植入术,术后每周至康复机构行言语康复治疗。目前患儿语言发育良好,智力发育评估同同龄儿,运动发育落后于同龄儿。
2 结 果
根据患儿典型临床表现及影像学检查(图1),诊断为Ⅳ型Waardenburg 综合征(WS 4),合并孤立肾。患儿5+月龄时行染色体核型分析检查,结果显示,G显带染色体核型分析9号臂间倒位(图2)。目前尚未完成基因测序,无法明确基因变异的具体情况。
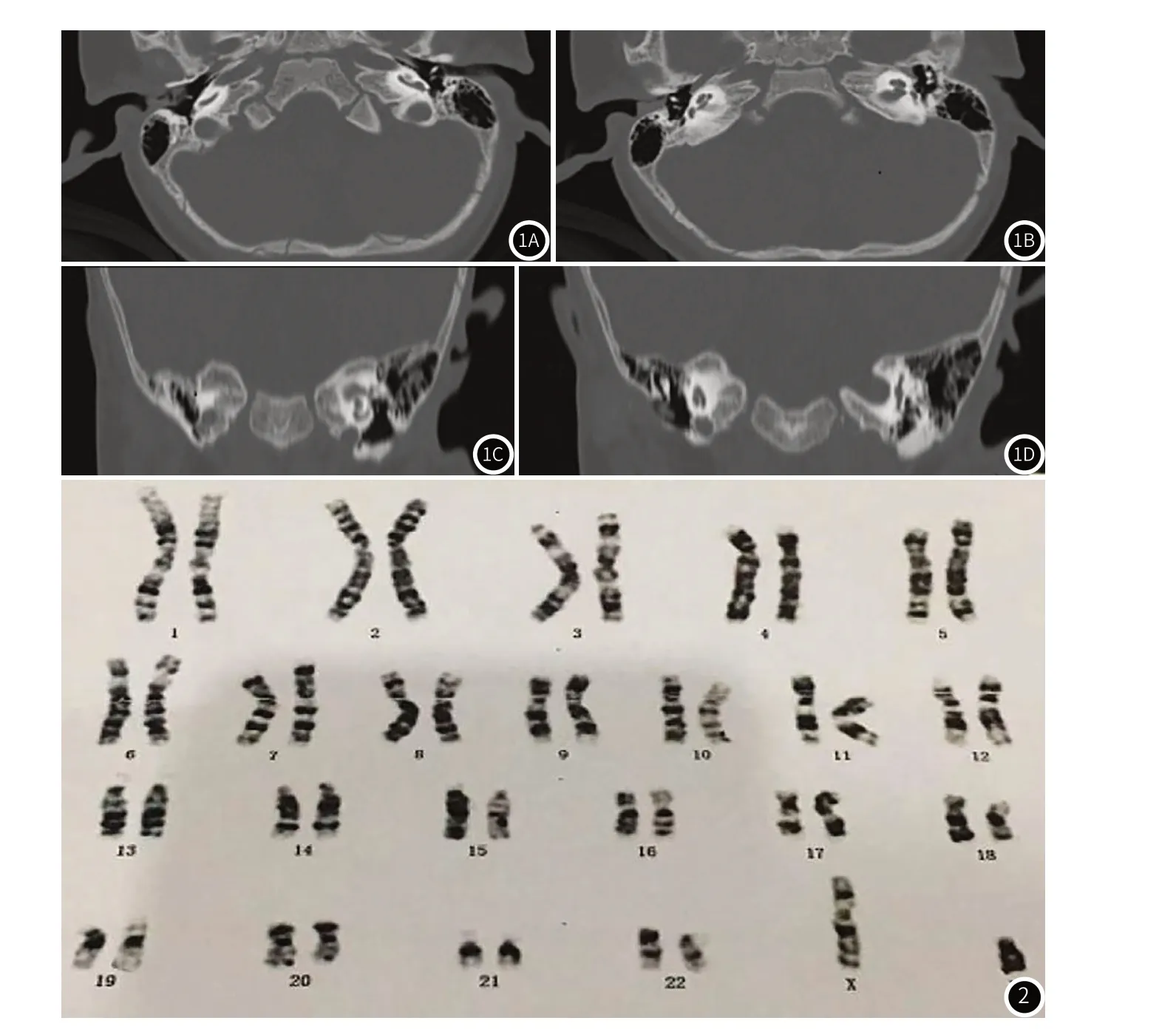
图1 患儿颞骨CT。图2 染色体核型分析图。
3 讨 论
3.1 WS的流行病学情况Waardenburg[1]首次报道WS时通过遗传学和统计学分析推测,WS在人群中发病率为1/42000。后续研究发现,WS发病率较Petrus推测的高。Nayak等[2]2003年的研究报道WS发病率为1/20000,Zaman等[3]在2014年报道的发病率为(0.119~0.208)/1000。聋人群体中WS发病率约0.9%~2.8%,先天性聋群体WS发病率为2%~5%[2]。杨淑芝等[4]在2 0 1 0 年的研究中报道的先天性聋儿的W S 发病率为0.69%。WS的发病率是否存在人种及性别差异,尚未见报道。Song等[5]在2016年进行了一项回顾性研究,纳入73篇文章,结果显示,71%的WS患者伴发耳聋,多为双侧极重度感应神经性耳聋,各亚型WS的耳聋发生率显著不同:WS1:52.3%;WS2:91.6%;WS3:57.1%;WS4:83.5%。
3.2 WS的诊断及分型WS具有高度的遗传异质性,临床表现也多种多样;根据不同的特征性临床表型将其分为四种亚型:Ⅰ型WS(WS1),Ⅱ型WS(WS2),Ⅲ型WS(WS3),Ⅳ型WS(WS4)。临床上常依据Waardenburg综合征协会推荐的诊断标准(表1)对WS1进行诊断,WS1的诊断必须满足至少两个主要诊断标准,或一个主要诊断标准加二个次要诊断标准。在WS1的诊断基础上做出其他3种亚型WS的诊断。与WS1相比,WS2无内眦移位,其他表型相同。而WS3在WS2表型基础上合并上肢畸形。WS4又称Waardenburg-Shah综合征或Waardenburg-Hirschsprung病,在WS2的基础上伴巨结肠病、胃肠道闭锁症。WS4还可能伴有神经病学特征:周围神经脱髓鞘病变(peripheral demyelinating neuropathy)、中枢性髓鞘形成障碍(central dysmyelinating leukodystrophy),这些类型被定义为PCWH(peripheraldemyelinating neuropathy, central dysmyelinating leukodystrophy,WS and HD)。已有的文献报道表面临床上以WS1及WS2常见,WS3及WS4少见[6]。

表1 Waarendburg综合征的诊断标准
3.3 WS合并肾脏异常情况WS与肾脏异常同时存在的病例很少见,最早的一例由Kaplan等[7]在1988年报道,表现为单侧重复肾畸形;第二例由Jankauskiene等[8]在1997年报道,为1例出生16d的女婴,表现为右多囊肾和左肾积水;随后Ekinci等[9]在2005年报道了第三例合并肾脏异常的病例,患儿为1岁女婴,表型为WS1合并双肾畸形(左肾收集系统重复伴左肾下级非梗阻性积水,右肾肾盂输尿管连接处梗阻)。Webb等[10]在2014年报道了一例WS合并肾缺如的病例,此患者家族中六代人出现了WS,少部分患者合并单肾缺如,作者提出WS合并肾缺如是否是一种新的WS亚型。除了WS合并肾脏畸形的报道,Anvesh等[11]在2018年也报道了WS2伴发肾病综合征的病例。本研究中的患儿WS4型诊断基本明确,合并单肾缺如,支持Webb等[10]的假设。神经嵴细胞发育缺陷及障碍是目前公认的WS可能的病因,而胚胎肾结构也受神经嵴细胞发育的影响,从而可以解释为什么部分WS患者会合并肾脏畸形,也提醒在遇到WS患者时,还应关注其肾脏的情况。
3.4 WS相关基因突变WS的遗传机制复杂,目前已被证实与WS相关的基因有PAX3、MITF、SNAI2、SOX10、EDNRB,EDN3[12]。前四个是转录因子,后两者是信号分子。各致病基因间存在相互作用,在神经嵴细胞的发育、迁徙及黑色素细胞的发育、迁徙中对其造成影响,导致各种表型WS的发生。但目前仍有一部分WS的致病基因不明确,有待进一步的研究。
PAX3(Paired Box 3,配对基因盒3)基因位于2号染色体q35-q37区间,编码PAX3蛋白,参与神经中枢、体节和骨骼肌发育以及神经嵴来源的心脏组织、黑色素细胞的发育。PAX3基因突变可以导致黑色素细胞合成异常、神经嵴发育异常。80%的WS1患者及50%的WS3患者可检测到PAX3基因突变[13]。已报道的与WS相关的PAX3变异多达100余种[14],国内报道了20余种[15]。PAX3基因突变多位于第2~6外显子[16]。Song等[5]报道显示与PAX3突变相关的WS患者中,50%的患者有听力损失,李进等[14]报道的合并听力异常的比例更高,10例WS患者中8例有听力损失。已有的文献报道中PAX3导致的WS1病例均为常染色显性遗传,WS3病例常染色体显性或隐性遗传情况均有存在。
小眼畸形相关转录因子(microphthalmia-associated transcription fctor,MITF )基因位于3号染色体12.3-14.1区间,编码的产物是黑色素细胞发育的关键转录因子,在生物体黑素细胞的迁徙、表达等方面起了一定的作用。MITF在WS的发病中体系复杂,总结为MITF基因突变,导致神经嵴来源的血管纹中间细胞缺失,从而影响耳蜗内淋巴液钾离子浓度,导致耳蜗内电位的消失,最终产生了耳聋。MITF基因变异主要导致WS2的发生,也有文献报道可导致WS4发生[17]。约15%~20%的WS2患者有MITF的变异[18]。已报道的MITF变异导致的WS2患者均为常染色体显性遗传病例。
SNAI2(Snail homologo 2)是Snail家族的一员,编码一种锌指转录因子,其位于8号染色体11.21。Sánchez-Martín等[19]在2002年对无MITF突变的38例WS2患者进行检测时发现,2例WS2表现为SNAI2纯合缺失。Pérez-Mancera等[20]在2007年的研究证实小鼠SNAI2杂合突变可引起小鼠额顶及腹部毛发变白,提示人类SNAI2的变异可能导致WS。但有关SNAI2与WS之间的关系有待进一步的研究。
SOX10基因[SRY(Sex determining region Y)-Box10,性别决定区盒基因]定位于22q13.1区,其编码的蛋白质是神经嵴发育的关键转录因子,在黑色素细胞和肠道神经节的发育中发挥重要作用。SOX10在内耳发育早期即广泛表达。Pingault等[21]在1998年首次报道了在WS4家系患者中发现SOX10杂合突变,明确了SOX10基因突变可导致WS4的发生。Bondurand等[22]在2007年首次报道在WS2病例检测发现SOX10缺失。文献统计约15%的WS2及50%的WS4与SOX10变异相关[23,11]。赵娜等[15]总结了SOX10变异共24例,其中17例为WS2患者,7例为WS4患者。SOX10变异导致的WS患者出现听力损失的情况更常见,程度更重;SOX10基因突变还能导致PCWH的发生,已有的研究也说明SOX10基因突变导致的WS4,患者常伴双侧内耳畸形。
内皮素3(Endothelin-3,EDN3)基因定位于20q13.2-13.3区间,内皮素受体 B(Endothelin receptor type B,EDNRB)基因定位于13q22。两者编码的产物相互作用,对神经嵴细胞的发育有重要影响。EDN3及EDNRB3基因突变主要与WS4相关,也有极少病例报道其突变引起WS2。已有的研究指出,20%~30%的WS4由此两种基因突变引起[12]。
3.5 WS的治疗Waardenburg综合征目前尚无有效的药物治疗。因大多数WS患者伴发严重的听力障碍,目前的目标是早期发现、早期干预,避免因耳聋造成言语发育等方面的影响。耳聋的主要治疗方式有佩戴助听器及植入人工耳蜗,已有的报道多表明人工耳蜗植入后效果良好,与其他类型耳聋相比无明显差别,也有部分WS患者植入后疗效欠佳[24]。WS的基因治疗依然在探索中,研究者们期待通过寻找到靶基因并对其做出修改,以达到治疗疾病的目的。
