柱前衍生-气相色谱-串联质谱法检测鹅肉中青霉素G残留量
2021-07-26谢星刘楚君陈晋元贺兆源卢阳王冉谢恺舟
谢星 刘楚君 陈晋元 贺兆源 卢阳 王冉 谢恺舟
摘要:建立了鹅肉中青霉素G残留的柱前衍生-气相色谱-串联质谱检测方法。鹅肉通过加速溶剂萃取(ASE 350),0.2 mol/L磷酸盐缓冲溶液(pH值8.0)提取,所得提取液过HLB(60 mg/3 mL)固相萃取柱净化,净化液经N2吹干后,乙醇复溶,三甲基硅烷基重氮甲烷(TMSD)衍生,所得衍生产物供气相色谱-串联质谱(GC-MS/MS)检测分析,色谱柱为TG-1MS(30.0 m×0.25 mm,0.25 μm),采用EI模式,全扫描(SCAN)和选择离子监测(SIM)方式定性,选择反应监测(Auto SRM)方式结合外标法定量。结果表明,鹅肉中青霉素G在4.50~100.00 μg/kg添加范围时,回收率为80.67%~91.02%,相对标准偏差(RSD)为1.57%~2.58%,日内RSD为2.72%~4.47%,日间RSD为3.51%~5.14%。鹅肉中青霉素G检测限为1.50 μg/kg,定量限为4.50 μg/kg,该方法灵敏度高,定性、定量准确,适用于鹅肉中青霉素G残留的确证检测。
关键词:鹅肉;青霉素G;残留;柱前衍生;气相色谱-串联质谱法
中图分类号:TS251.7 文献标志码: A
文章编号:1002-1302(2021)11-0132-06
收稿日期:2020-09-06
基金项目:国家现代农业产业技术体系建设专项(编号:CARS-41-G23);江苏高校优势学科建设工程资助项目(编号:PAPD)。
作者简介:谢 星(1989—),女,江苏扬州人,博士,助理研究员,主要从事动物产品安全研究。Email:yzxx1989@163.com。
通信作者:谢恺舟,博士,教授,主要从事动物产品品质、安全与兽药残留检测方法研究。E-mail:yzxkz168@163.com。
青霉素G主要作用于革兰氏阳性菌,是临床上常用的一类抗生素。对家禽而言,青霉素G可以预防敏感菌所致的全身(局部)感染 [1],应用过程中常存在使用不合理和不规范现象,导致禽肉中青霉素G残留量超标,最终危害人体健康。因此,各国对动物性组织中青霉素G残留限量制定了严格标准,其中,欧盟、日本、加拿大和中国农业农村部规定鸡组织中青霉素G的最高残留限量(maximum residue limit,MRL)均为50 μg/kg [2-5]。本研究参考上述MRL标准建立鹅肉中青霉素G残留的确证检测方法。
目前,国内外关于检测动物性食品中青霉素G的方法虽然已有微生物法 [6]、免疫分析法 [7]、薄层色谱法 [8]、高效液相色谱-紫外检测法(HPLC-UV) [9]、高效液相色谱-串联质谱法(HPLC-MS/MS) [10]等,但使用气相色谱-串联质谱法(GC-MS/MS)检测动物性组织中青霉素G残留的方法还未有报道。与其他方法相比,柱前衍生-气相色谱-串联质谱法(GC-MS/MS)灵敏度高、抗干扰能力强、前处理简单和快速。因此,本研究旨在建立GC-MS/MS检测鹅肉中青霉素G残留的确证分析方法,为动物源性食品中青霉素G残留检测标准的制定提供技术支持。
1 材料与方法
1.1 仪器与试剂
气相色谱-串联质谱仪(配有Trace 1300型气相色谱仪、TSQ 8000型三重四级杆串联质谱仪、Triplus RSH自动进样器)(美国Thermo Fisher公司)、加速溶剂萃取仪(ASE 350型,美国Thermo Fisher公司);电子分析天平(AE260S型,瑞士Mettler Toledo公司);漩涡混合器(G560E型,美国Scientific Industries有限公司);全自动多管涡旋振荡器(TBOYS型,美国Troemner有限责任公司);烘箱(FD115型,德国Binder公司);氮吹仪(N-EVAP-112型,美国Organomation公司);超纯水制备仪[Smart2-Pure型,賽默飞世尔科技(中国)有限公司];实验室pH计[FE20型,梅特勒-托利多仪器(上海)有限公司]等。
青霉素G钾标准品(纯度≥98%,CAS号为113-98-4,美国SIGMA-ALDRICH有限公司);三甲基硅烷基重氮甲烷[TMSD,CAS号为18107-18-1,阿拉丁试剂(上海)有限公司];乙腈、甲醇(色谱纯,美国Merck有限公司);乙醇(色谱纯,美国Fisher公司);十二水合磷酸氢二钠、磷酸二氢钾、氢氧化钠、正己烷(分析纯,国药集团化学试剂有限公司);超纯水 [电阻率为18.2 MΩ·cm(25 ℃),符合国家实验室用水标准(GB 6682—1992)]。
1.2 主要溶液的配制
准确称取青霉素G标准品10.20 mg(纯度为98%)置于10 mL的棕色容量瓶中,用乙醇溶解并定容至刻度,摇匀,配成质量浓度为1.00 mg/mL的标准品储备液,并将标准品储备液用乙醇分别准确逐级稀释成100.0、10.0、1.0、0.1 μg/mL标准工作液,置于4 ℃冰箱中保存备用。
0.2 mol/L磷酸盐缓冲溶液:准确称取67.7 g十二水合磷酸氢二钠(Na2HPO4·12H2O)和1.5 g磷酸二氢钾(KH2PO4),采用超纯水溶解并定容至1 000 mL,配成0.2 mol/L pH值8.0的磷酸盐缓冲溶液。
10%氢氧化钠溶液:准确称取氢氧化钠固体1.0 g,用超纯水溶解并定容至10 mL,配成10 mg/g氢氧化钠溶液。
1%甲醇乙腈溶液:量取5 mL甲醇于500 mL容量瓶中,用乙腈定容至刻度,摇匀,配成1%甲醇乙腈溶液。
80%乙腈:量取20 mL水于100 mL容量瓶中,用乙腈定容至刻度,摇匀,配成80%乙腈溶液。
1.3 试验方法
1.3.1 试验鹅饲养与样品采集
随机选取70日龄扬州鹅(扬州天歌鹅业发展有限公司)公鹅10羽、母鹅10羽,单笼饲养,试验期间均饲喂不添加任何药物的全价饲料(扬州市扬大饲料厂提供),自由饮水。饲养14日后屠宰,采集试验鹅胸大肌肌肉作为空白样品,分装并密封,置于-34 ℃冰箱中保存备用。
1.3.2 样品提取
准确称取(2.0±0.02) g均质好的空白样品和4.0 g硅藻土置于研钵中,将其研磨成小颗粒,填入22 mL萃取池,萃取池置于ASE 350进行提取,设置参数,压力:10.34 MPa;加热温度:30 ℃;静态提取时间:5 min;冲洗溶剂总量:40%;每个样品之间自动冲洗1次;氮气吹扫时间:60 s;首先用正己烷去除样品中的脂肪,提取1次,弃掉提取液;其次用0.2 mol/L磷酸盐缓冲溶液(pH值8.0)提取样品中的目标物,提取2次,收集提取液,待用。
1.3.3 样品净化与浓缩
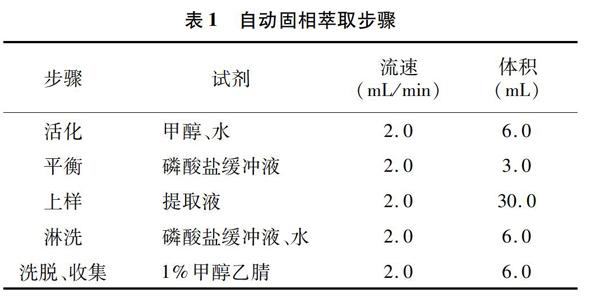
将“1.3.2”节中收集的提取液经过HLB固相萃取柱(60 mg/3 mL)净化,自动固相萃取步骤见表1。收集的洗脱液置于氮吹仪中35 ℃吹干,待用。
1.3.4 样品复溶与衍生化
将“1.3.3”节中氮吹至干的净化收集液加入100 μL乙醇溶解,涡旋混匀1 min,然后加入400 μL TMSD,密封,于30 ℃烘箱中避光反应30 min后取出,乙醇定容至1.0 mL,然后转移至2.0 mL具塞离心管中,涡旋混匀1 min,常温下12 000 r/min离心10 min,经过0.22 μm有机相针头式滤器过滤,滤液供GC-MS/MS检测。
1.3.5 气相色谱与质谱条件
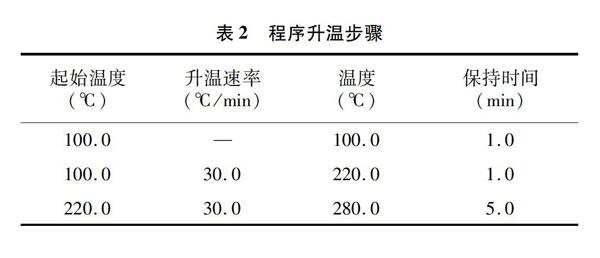
毛细管色谱柱:TG-1MS(30.0 m×0.25 mm,0.25 μm,美国Thermo Fisher公司);载气:高纯氦气(>99.999%,413.7 kPa),流速:1.0 mL/min。進样口温度:280 ℃;分流模式:不分流进样;分流流量:50.0 mL/min;不分流时间:1.0 min;载气模式:恒流模式;载气流速:1.0 mL/min;2 min后开阀,载气节省时间2 min,载气节省流量20.0 mL/min;进样体积:1.0 μL。程序升温步骤见表2。
电离模式:电子轰击离子源(EI);电子束能量(电离能):70 eV;碰撞气:高纯氩气(>99.999%,275.8 KPa);离子源温度:280 ℃;传输线温度:280 ℃;溶剂延迟:5.0 min;采集数据模式:全扫描(SCAN)和选择离子监测(SIM)方式定性,选择反应监测(Auto SRM)方式定量。
1.3.6 标准曲线绘制
按“1.3.2”节处理方法制备空白鹅肉基质提取液,分别移取适量的空白基质提取逐级稀释青霉素G的标准工作液,使青霉素G的添加浓度为定量限(LOQ)、15.0、25.0、50.0、100.0、150.0、200.0 μg/kg。将各浓度样品按“1.3.3”节、“1.3.4”节样品前处理方式进行GC-MS/MS检测分析,每个浓度重复测定6次,取平均值。以青霉素G标准工作液在空白基质中添加浓度为横坐标(x),以青霉素G衍生产物的定量离子对m/z 174.1>114.1*的峰面积为纵坐标(y),绘制基质标准曲线并作为待测样品的定量曲线。
1.3.7 样品加标回收率和精密度测定
准确称取2.0 g均质好的空白鹅肌肉样品,按“1.3.2”节的方法将空白样品与硅藻土充分研磨均匀,加入青霉素G标准工作液适量,使其最终在每个空白样品中的添加浓度为LOQ、0.5 MRL、1.0 MRL和2.0 MRL,每个添加浓度设6个平行,按“1.3.3”节、“1.3.4”节的方法处理进行GC-MS/MS检测,最终将检测结果带入空白基质标准曲线中求得浓度,计算样品加标回收率。将LOQ、0.5 MRL、1.0 MRL和2.0 MRL浓度样品在1 d内不同时间用同一标准曲线和1周内不同天和不同标准曲线用均用同一台GC-MS/MS重复测定6次,分别计算日内和日间精密度。
1.3.8 灵敏度测定
采用空白基质提取液逐级稀释低浓度的青霉素G标准工作液,用已建立的GC-MS/MS 方法进行检测。每个浓度进行6次重复测定,计算平均信噪比(S/N)。当S/N≥3时,所对应的青霉素G浓度作为该方法的检测限(LOD);当S/N≥10时,所对应的青霉素G浓度作为该方法的定量限(LOQ)。
2 结果与分析
2.1 母离子和子离子的确定
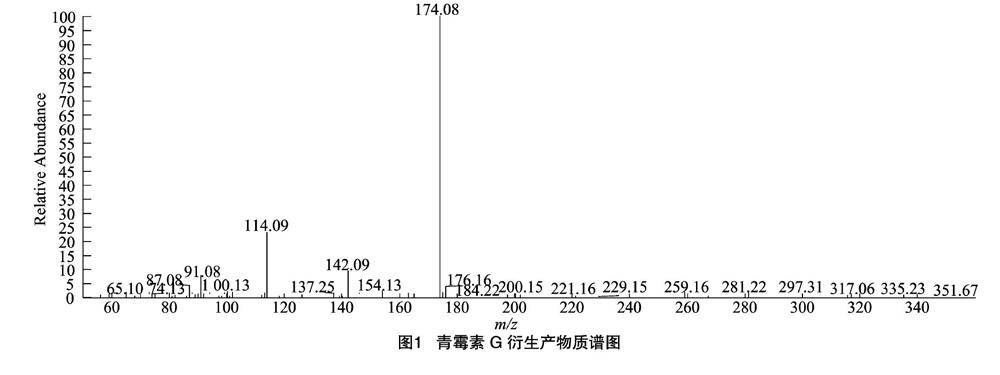
准确吸取青霉素G与TMSD衍生后的标准溶液(100.0 μg/mL)用电子轰击离子源(EI)进行全扫描(Full SCAN)和选择离子扫描(SIM),获得衍生产物的全扫描色谱图和质谱图,根据质谱图分析特征离子结构,同时,参照Preu等提出的青霉素G与重氮甲烷反应的质谱图结构和梯度试验 [11],挑选质荷比(m/z)比较大且丰度比较高的母离子,最终得到衍生产物的质谱图。由图1可知,青霉素G衍生产物的母离子是m/z 174.1。
确定好母离子(即定量离子)后,就需考虑子离子(即定性离子)的选择。采用Auto SRM优化仪器方法,查看母离子在不同碰撞能下产生的子离子碎片强度信息,选择合适的碰撞能和较大强度的碎片离子作为子离子。由母离子与子离子组合为监测离子对。为对青霉素G衍生产物进行准确的定性、定量,本研究选择3个响应强度较高且稳定存在的监测离子对。青霉素G三甲基硅甲酯的保留时间和相关质谱参数见表3。
2.2 色谱图
空白鹅肉样品总离子流色谱图(TIC)和定量、定性离子的质量色谱图(MC)及空白鹅肉添加50.0 μg/kg 青霉素G标准品的总离子流色谱图(TIC)和定量、定性离子的质谱图(MC)。由图2、图3可知,在优化的样品前处理和GC-MS/MS条件下,青霉素G与TMSD反应的衍生产物与杂质良好地分离,峰形正常良好,保留时间在10.85 min。
2.3 基质标准曲线的绘制
以青霉素G标准工作液在不同空白基质中添加浓度为横坐标(x),青霉素G衍生产物的定量离子对m/z 174.1 > 114.1*的峰面积为纵坐标(y),绘制基质标准曲线(图4)。由图4可知,青霉素G线性范围为4.50~200.0 μg/kg时,回归方程为y=27 567x-38 053,决定系数r2为0.999 6。
2.4 添加回收率和精密度
青霉素G在空白鹅肌肉的添加浓度为LOQ、0.5 MRL、1.0 MRL和2.0 MRL时,青霉素G的添加回收率及精密度见表4。由表4可知,青霉素G在鹅肌肉中的添加浓度范围在LOQ~2.0 MRL之间时,青霉素G在鹅肌肉中的添加回收率为80.67%~91.02%,相对标准偏差(RSD)为1.57%~2.58%,日内RSD为2.72%~4.47%,日间RSD为3.51%~5.14%。
2.5 检测限与定量限
经灵敏度测定,鹅肌肉中青霉素G的检测限(LOD)为1.50 μg/kg、定量限(LOQ)为4.50 μg/kg。
3 讨论
3.1 毛细管色谱柱的选择
由于青霉素G属于β-内酰胺类抗生素,其β-内酰胺环的羰基α-碳上有一个酰胺基侧链,而在它稠和的环上有1个2位-羧基 [12],故青霉素G属于强极性化合物。因为GC-MS/MS只能检测极性低、沸点低的化合物,所以不能直接检测青霉素G,需将其进行衍生化反应,降低其极性,才能用GC-MS/MS检测。由于不知衍生产物的极性大小,所以本试验采用盲摸原则(色谱柱极性由弱到强)和固定相相似性原则(固定相与待测物极性的相似性)进行操作。试验初期先采用实验室现有的非极性毛细管色谱柱TG-5MS(30.0 m×0.25 mm×0.25 μm,5%-二苯基-95%-二甲基硅氧烷),发现无法检测到未知衍生物,然后又参考Preu等在GC-MS检测牛肉中的青霉素G时,选用Rtx-1(30.0 m×0.25 mm,0.25 μm)非極性毛细管色谱柱 [11],结果青霉素G峰形良好。综合来看,青霉素G衍生物极性很弱,需选用比TG-5MS极性还要低的毛细管色谱柱。本试验最终选择TG-1MS(30.0 m×0.25 mm,0.25 μm,100%聚二甲基硅氧烷)毛细管色谱柱检测鹅肉中的青霉素G残留,发现青霉素G峰形更好,与Preu等报道的结果 [11]一致。这是因为TG-1MS为完全非极性毛细管色谱柱,惰性更高,可保证良好峰形和灵敏度,适用于难以分析的化合物;可降低基线噪声,提高信噪比;耐受温度可达330 ℃或350 ℃,高温操作时也不会出现柱流失。
3.2 衍生化试剂的选择与衍生产物的稳定性
衍生化试剂种类很多,包括甲硅烷基化试剂、酰化试剂、烷基化试剂及酯化试剂等。重氮甲烷是烷基化试剂中常用的甲基化试剂,主要将羧酸转化为甲酯(重氮链),然后通过GC-MS/MS进行测定。Preu等 [11]、伊冰 [13]都曾用重氮甲烷衍生青霉素G,前者采用的是GC-MS结合内标法,结果良好,后者是GC结合外标法,结果较差。虽然重氮甲烷与羧酸类化合物反应迅速,但需现用现配,否则会在色谱图中产生杂峰,且不易制备和分解较快,同时具有高毒性、热不稳定性和易爆炸性等性质 [14]。本试验也对此进行验证,最终效果不理想,得出重氮甲烷并不是实验室常用衍生试剂的最佳选择,需要找一个更安全、稳定的替代试剂。三甲基硅烷基重氮甲烷(TMSD)与重氮甲烷具有相同的衍生能力,而且TMSD具有无毒、安全、稳定的性质,更方便合成药物用于分析,也更适用于实验室操作。TMSD主要是通过用三甲基甲硅烷基取代一个氢来克服重氮甲烷的缺点 [15-16]。虽然TMSD是重氮甲烷的安全替代品,但用TMSD作为青霉素G的衍生化试剂目前尚未见报道,因此需验证TMSD能否与青霉素G发生反应。本试验首先将TMSD、青霉素G及二者的反应产物进行薄层色谱点板并在紫外分析仪下照射,观察是否发生反应,结果发现衍生产物的点处于最前端,证明有反应发生,然后用GC-MS/MS 测定分析;同时参考Presser等 [16]、Ranz等 [17]、Rompa等 [18]采用TMSD衍生羧酸类物质并用GC进行分析检测的方法,试验结果良好。故本试验最终采用GC-MS/MS结合外标法检测鹅肉中青霉素G残留,选择TMSD作为衍生化试剂,不需购买内标物,降低试验成本,无需现用现配,保存周期长且不易分解,缩短样品前处理过程,结果良好。综上所述,本试验选用TMSD作为衍生化试剂是最佳选择。此外,本试验对青霉素G衍生产物在24 h内的稳定性进行了研究,由图5可知,整体呈现稳定趋势,但8~12 h波动较大,可能与其反应是否充分有关,然后从12 h往后青霉素G衍生产物开始分解。因此,青霉素G衍生产物最好能在24 h内完成检测分析。
3.3 气相色谱和质谱参数的优化
本试验参照Preu等建立的方法 [11],所得目标物的峰形不好,且出峰时间较长,约21.08 min,因此需要优化气相色谱参数。通过反复优化试验,所得目标物的峰形较好,且出峰时间大大缩短,最终筛选出气相色谱最佳条件为:初始温度100 ℃,1 min;
30 ℃/min升至220 ℃,1 min;30 ℃/min升至280 ℃,5 min。进样口温度:280 ℃;不分流进样模式;不分流时间:1.0 min;50.0 mL/min的分流流量;恒流模式;载气流速1.0 mL/min;2 min后开阀,载气节省2 min,载气节省20.0 mL/min流量;进样体积1.0 μL。
众所周知,未知化合物的定性主要是通过将目标化合物的标准品Full SCAN获得的质谱图与Nist谱库比对。由于青霉素G衍生产物质谱图在谱库中不存在,所以主要根据Preu等提出的质谱图和大量的预试验确定的目标物来进行优化试验 [11]。故本试验最终采用Full SCAN和SIM方式对青霉素G衍生物进行定性,扫描范围m/z 50~360(图1);TSQ 8000采用Auto SRM方式进行定量扫描,选择m/z 174.1>91.1和m/z 174.1>142.1为定性离子对,m/z 174.1>114.1*为定量离子对,确保定性和定量结果的准确性。
4 結论
本试验优化并建立了鹅肉中青霉素G残留的GC-MS/MS检测方法。鹅肉中青霉素G回收率高于80.67%,LOD、LOQ分别为1.50、4.50 μg/kg。该检测方法灵敏度高、定性、定量准确,可满足鹅肉中青霉素G残留确证检测的要求。
参考文献:
[1]Iliadis N,Petridou E N,Foukos A. Clinical and subclinical bovine mastitis in area of Kilkis [J]. Journal of the Hellenic Veterinary Medical Society,2018,48(1):32.
[2]The European Medicines Agency. Commission regulation (EU) No. 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin[S]. 2010.
[3]The Japan Food Chemical Research Foundation. 2015 [Z]. 2015.
[4]Canada H. List of maximum residue limits(MRLs)for veterinary drugs in foods [Z]. 2018.
[5]中华人民共和国农业农村部,国家卫生健康委员会,国家市场监督管理总局.食品安全国家标准 食品中兽药最大残留限量:GB 31650—2019[S]. 2019.
[6]Reynoso E,Nesci A,Allegretti P,et al. Kinetic and mechanistic aspects of sensitized photodegradation of β-lactam antibiotics:microbiological implications [J]. Redox Report:Communications in Free Radical Research,2012,17(6):275-283.
[7]He X,Duan C F,Qi Y H,et al. Virtual mutation and directional evolution of anti-amoxicillin ScFv antibody for immunoassay of penicillins in milk [J]. Analytical Biochemistry,2017,517:9-17.
[8]Choma I. Thin-layer chromatography hyphenated with bioassays [J]. Journal of AOAC International,2013,96(6):1165-1166.
[9]Castillo-García M L,Aguilar-Caballos M P,Gómez-Hens A. Determination of veterinary penicillin antibiotics by fast high-resolution liquid chromatography and luminescence detection [J]. Talanta,2017,170:343-349.
[10]Huang Z,Pan X D,Huang B F,et al. Determination of 15 β-lactam antibiotics in pork muscle by matrix solid-phase dispersion extraction (MSPD) and ultra-high pressure liquid chromatography tandem mass spectrometry [J]. Food Control,2016,66:145-150.
[11]Preu M,Petz M. Isotope dilution GC-MS of benzyl penicillin residues in bovine muscle [J]. The Analyst,1998,123(12):2785-2788.
[12]周 杰,赵 静,董 超,等. 分散固相萃取结合UPLC-MS/MS测定柑橘中青霉素G及其代谢物残留 [J]. 分析测试学报,2019,38(4):442-448.
[13]伊 冰. 测定动物性食品中7种青霉素残留物的气相色谱法 [J]. 国外医学:卫生学分册,1992(3):189-190.
[14]Dallinger D,Kappe C O. Recent enabling technologies for diazomethane Generation and reactions [J]. Aldrichimica Acta,2016,49(3):57-66.
[15]Chhonker Y S,Haney S L,Matthiesen R A,et al. Quantitative determination of a potent geranylgeranyl diphosphate synthase inhibitor using LC-MS/MS:derivatization and application [J]. Journal of Pharmaceutical and Biomedical Analysis,2018,153:22-28.
[16]Presser A,Antje H. Trimethylsilyl diazomethane-A mild and efficient reagent for the methylation of carboxylic acids and alcohols in natural products [J]. Monatshefte für Chemie,2004,135(8):1015-1022.
[17]Ranz A,Korpecka J,Lankmayr E. Optimized derivatization of acidic herbicides with trimethylsilyldiazomethane for GC analysis [J]. Journal of Separation Science,2008,31(4):746-752.
[18]Rompa M,Kremer E,Zygmunt B. Derivatisation in gas chromatographic determination of acidic herbicides in aqueous environmental samples [J]. Analytical and Bioanalytical Chemistry,2003,377(4):590-599.
