LC-MS/MS法测定食用植物油中 乙基麦芽酚的不确定度评定
2021-07-26罗小宝周芳梅
◎ 罗小宝,孙 艺,周芳梅,陈 茹
(1.广东省食品工业研究所有限公司,广东 广州 511442;2.广东省食品工业公共实验室,广东 广州 511442;3.广东省食品质量监督检验站,广东 广州 511442)
乙基麦芽酚常在食品香精的调香过程中被广泛应用,但在《食品安全国家标准 食品添加剂使用标准》(GB 2760—2014)[1]附录B中规定了食用植物油中不得添加食用香精。乙基麦芽酚香味极强,少量即可达到增香的目的。目前,各地政府公布的不合格公告中有不少不法商家在食用植物油中掺入乙基麦芽酚,将不合格产品卖给消费者。乙基麦芽酚具有强还原性,能够与金属形成螯合物,影响人体内铁和铝含量的水平。为了人体的健康和安全的需要,有必要进行严控[2]。
本研究对食用植物油中乙基麦芽酚测定过程中不确定度的来源进行分析,建立来源图和数学模型量化不确定度各分量,全面分析不确定度来源并进行计算合成标准不确定度和扩展不确定度,以确保检测结果的准确可靠[3-8]。
1 材料与方法
1.1 试剂与仪器
甲醇(色谱纯,上海安谱);甲酸(色谱纯,上海安谱);乙基麦芽酚(标准物质,CAS号为4940-11-8,美国Sigma公司)。
ACQUITY H-Class PLUS/XevoTQ-S micro液相色谱-串联质谱仪(美国Waters公司),配有电喷雾离子源;AR323CN分析天平(奥豪斯);超纯水机(密理博)。
1.2 试验方法
依据标准《食用植物油中乙基麦芽酚的测定》(BJS 201708)进行测定。准确称取10 g试样(精确至0.001 g)于50 mL的离心管中,并用移液枪准确移入10.0 mL的甲醇,振摇3 min后,在离心机于4 ℃条件下用10 000 r·min-1离心10 min,移取上清液于25 mL的具塞刻度管中,再用10.0 mL甲醇分两次提取,合并所有上清液到刻度管中,用甲醇定容至20.0 mL,经0.22 μm有机相滤膜过滤,等上机分析。
1.3 标准曲线的绘制
(1)乙基麦芽酚标准储备液。准确称取0.010 07 g乙基麦芽酚于10 mL容量瓶中并用甲醇溶解至刻度,该浓度为 1.007 mg·mL-1。
(2)标准中间液。准确移取1.00 mL储备液于10 mL容量瓶中,用甲醇定容,该浓度为100.7 μg·mL-1。
(3)标准工作液。准确分别移取中间液0.012 5 mL、0.025 mL、0.05 mL、0.25 mL和 0.50 mL于 1.00 mL的容量瓶中,用甲醇稀释至刻度配制出浓度分别为 1.259 μg·mL-1、2.518 μg·mL-1、5.035 μg·mL-1、25.18 μg·mL-1和 50.35 μg·mL-1的工作液,再分别准确移取0.200 mL标准工作液加入与试样基质对应的阴性试样中进行提取,与试样提取时间保持一致,制成的标准溶液的浓度分别为 12.59 ng·mL-1、25.18 ng·mL-1、50.35 ng·mL-1、251.8 ng·mL-1和 503.5 ng·mL-1。
2 数学模型
液相色谱-质谱/质谱法测定试样中乙基麦芽酚含量按式(1)计算。

式(1)中,X-试样中乙基麦芽酚的含量,μg·kg-1;C-试样中乙基麦芽酚的浓度含量,ng·mL-1;V-试样的定容体积,mL;m-试样的质量,g。
3 不确定度的来源分析
从测定原理、分析方法、数学模型等方面进行分析,LC-MS/MS测定食用植物油中乙基麦芽酚残留量的不确定度来源于标准品溶液配制、定容体积、样品称取、样品重复性和标准曲线等产生的不确定度[9-14]。具体相关不确定度主要来源见图1。
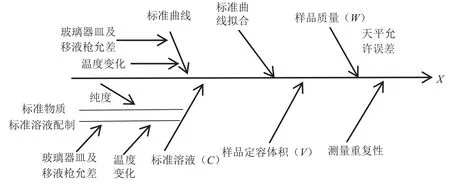
图1 LC-MS/MS法测定食用植物油中乙基麦芽酚的不确定度来源图
4 标准不确定度计算
4.1 配制标准储备液产生的相对合成标准不确定度urel,1(S)
4.1.1 标准物质引入的相对标准不确定度urel,2(P)
查标物证书可知,扩展不确定度为2.0%(k=2),纯度为99.9%,则其相对标准不确定度平方为:

4.1.2 称量引入的相对标准不确定度urel,3(m)
乙基麦芽酚标准物质由十万分之一电子天平称量。根据电子天平检定证书,最大允许误差(MPE)为±0.05 mg,按矩形分布计算,称取乙基麦芽酚标准物质10.07 mg,其相对标准不确定度平方为:

4.1.3 移液枪和容量瓶体积引入的相对标准不确定度urel,4(V1)
根据《常用玻璃量器检定规程》(JJG 196—2006),10 mL A级容量瓶的最大容量允差(MPE)为±0.020 mL,1 000 μL移液枪的最大容量允差(MPE)为±10.0 μL,根据欧洲分析化学中心(EURACHEM)认为其服从三角分布,则配制储备液产生的相对标准不确定度平方为:

配制中间液,使用1 000 μL的移液枪,取1 000 μL储备液,甲醇定容至10 mL容量瓶,按照三角分布,其产生的相对标准不确定度平方为:

移液枪和容量瓶体积引入的相对标准不确定度平方为:

4.1.4 温度系数引入的相对标准不确定度urel,8(T1)
在温度(20±4)℃条件下进行检测,引入的不确定估算是用该温度范围和体积膨胀系数进行计算,因为液体的体积膨胀明显大于玻璃容器的体积膨胀,所以只考虑液体的体积膨胀。甲醇的膨胀系数为1.19×10-3/℃,按照矩形分布,温度系数引入的相对不确定平方为:

配制乙基麦芽酚标准储备液及中间液温度系数引入的相对标准不确定度平方为:

4.2 标准工作溶液配制过程中所用器量引入的相对标准不确度urel,12(V2)
标准工作溶液配制过程中所用器量引入的相对标准不确定度主要来源为容量瓶定容体积、移液枪移取溶液。
4.2.1 容量瓶定容体积引入的不确定度
配制用容量瓶(5个)经鉴定均为A级,根据《常用玻璃量器检定规程》(JJG 196—2006),1 mL单标线A级容量瓶的最大允差为±0.010 mL,按三角分布计算,相对标准不确定度的平方为:

4.2.2 移液器引入的不确定度

表1 使用移液枪引入的相对标准不确定度平方表
标准工作溶液配制过程中所用器量带来相对标准不确定度的平方:

4.3 温度系数引入的相对标准不确定度分urel,13(T2)
溶液温度与检定标准温度(20 ℃)不同引入的相对标准不确定度分量urel,13(T2),在(20±4)℃条件下进行,甲醇的膨胀系数为1.19×10-3℃-1,以按照矩形分布计算,配制标准工作曲线时温度变化引入的相对标准不确定度见表2。

表2 温度变化引入的相对标准不确定度平方表
配制标准工作曲线时温度变化引入的相对标准不确定度的平方为:

配制乙基麦芽酚标准溶液及标准曲线引入的相对不确定度的平方:

4.4 标准曲线拟合产生的相对不确定度及样品重复性测定产生的相对标准不确定度urel,14(C)
4.4.1 测量结果重复性偏差引入的不确定度分量u(X)
选用一个不合格的试样,选取6份,经处理后,进行测定,测定结果见表3。

表3 样品重复性测定结果表(单位:ng·mL-1)
阳性样品中乙基麦芽酚重复性测定引入的标准不确定度按式(2)计算。

式(2)中,u2(X)-试样中乙基麦芽酚重复性测定引入的标准不确定度;xi-试样中乙基麦芽酚各重复测定的浓度含量的值,ng·mL-1;试样中乙基麦芽酚重复测定的浓度含量的平均值,ng·mL-1;i-每次测试;n-测试总次数。
4.4.2 标准曲线拟合的标准偏差引入的相对标准不确定度
将乙基麦芽酚标准使用液标准系列溶液测定3次,得到峰面积,取平均值,以乙基麦芽酚标准系列溶液浓度为x坐标,乙基麦芽酚标准系列溶液峰面积作为y坐标,用最小二乘法拟合求出一元回归方程及相关系数,计算结果见表4。
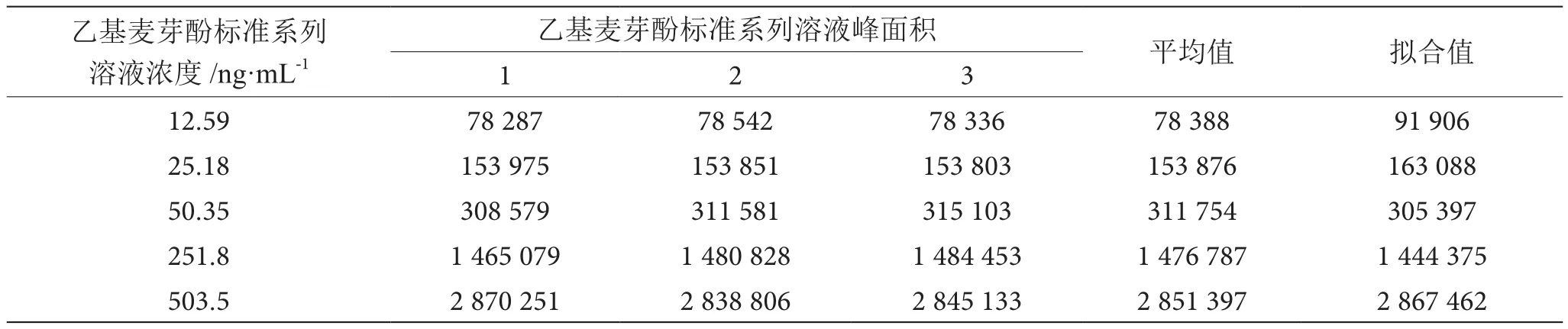
表4 乙基麦芽酚标准系列溶液3次测定的结果表
采用最小乘法拟合浓度-峰面积之比的标准工作曲线,乙基麦芽酚线性回归方程为y=5 653.9x+20 723,相关系数r=0.999,大于0.995,符合GB/T 27404—2008标准要求,表明乙基麦芽酚的标准系列溶液峰面积和标准系列溶液浓度具有显著的线相关性。根据贝塞尔公式,标准曲线的剩余标准差SR按式(3)计算。

式(3)中,yij-仪器各点峰面积;yi-回归直线计算值;n-测量点数,n=5;m-每个测量点重复测量次数,m=3。
由标准差求得,标准曲线拟合的标准偏差引入的相对标准不确定度u2(Y)。

回归系数斜率的不确定度u(a)。
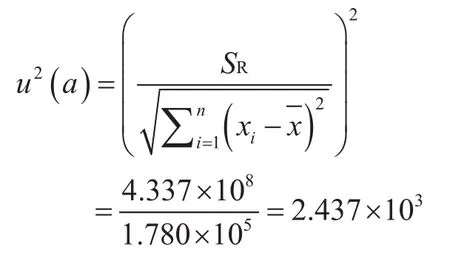
回归系数斜率的不确定度u(b)。

分量之间相互独立不相关,根据公式(4)计算。

按照不确定度传播律进行合成,需先得出传播系数,对线性方程y=ax+b,求偏导得到如下结果。
乙基麦芽酚各分量灵敏系数为。

合成标准不确定度uc(c)。

合成相对标准不确定度为:

4.5 样品处理过程中产生的相对不确定度urel,15(Q)
4.5.1 样品处理过程中称量引入的相对标准不确定度urel,16(m)
根据称量的千分之一电子天平使用说明书,称量10.000 g时,最大允差为±5 mg,按矩形分布用该天平秤量样品6份,质量分别为10 011 mg、10 026 mg、10 018 mg、10 023 mg、10 018 mg 和10 032 mg,方差为54 mg,所用校准砝码的引入的不确定度贡献较小,忽略不计。称取样品产生的相对标准不确定度的平方为:
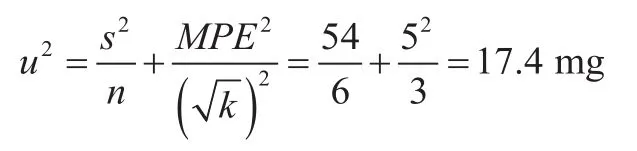
称取样品质量为10.005 g,10.033 g,其平均值为10.019 g,产生的相对标准不确定度平方为:

4.5.2 样品处理过程中所用器量引入相对标准不确定度urel,17(V3)
使用10 mL移液枪移取提取液,移取2次,其最大允差为±0.06 mL,依据三角分布,其相对标准不确定度平方为:

由于乙基麦芽酚含量较大,稀释2倍,取500 μL样液并加空白基质液进行稀释定容至1 mL容量瓶,使用的1 000 μL移液枪,最大允差为±10.00 μL,1 mL单标线容量瓶A级最大允差为±0.010 mL,则稀释过程产生的相对标准不确定度为:

4.5.3 温度系数引入的标准不确定度分量urel,21(T3)
溶液温度与检定标准温度(20 ℃)不同引入的相对标准不确定度分量μrel,21(T3),样品在(20±4)℃条件下进行定容,样品定容溶液为甲醇,甲醇的膨胀系数为1.19×10-3 ℃-1,按照矩形分布计算其相对标准不确定度平方为:
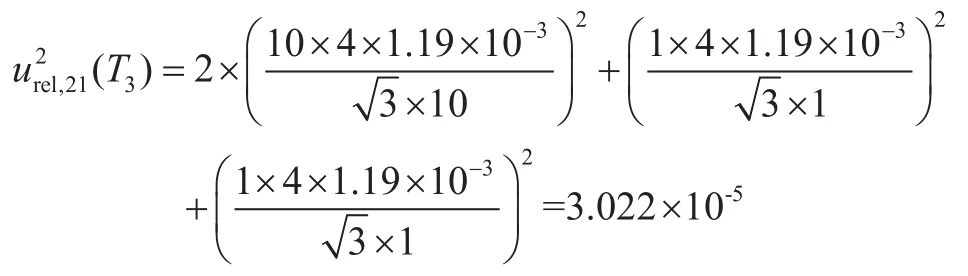
4.5.4 样品处理过程中引入的相对标准不确定度urel,15(Q)
样品处理过程引入的相对标准不确定度平方为:

5 合成相对标准不确定度的评定
根据含量计算公式,合成相对扩展不确定度由标准品溶液配制、定容体积、样品称取、样品重复测定以及最小二乘法拟合标准曲线等产生的不确定度分量组成,如表5所示,因此LC-MS/MS法测定食用植物油中乙基麦芽酚含量测定的合成相对扩展不确定为:


表5 食用植物油中乙基麦芽酚含量测定的合成相对标准不确定度分量表
5.1 扩展不确定度评定
相对扩展不确定度Urel=k×urel,假设该测量误差属于正态分布,包含因子k=2,取置信水平p=95%,则相对扩展不确定度为:Urel=2×3.61%=7.2%。
样品乙基麦芽酚检出190 μg·kg-1,其扩展不确定度为:U=190×7.2%=14 μg·kg-1。
5.2 测量不确定度的结果报告
根据不确定度评估后,乙基麦芽酚检出结果为190 μg·kg-1,U=14 μg·kg-1,k=2。
6 结论
通过对高效液相串联质谱法对植物食用油中乙基麦芽酚的不确定度的分析,得出测定过程中不确定度来源主要有由标准品溶液配制、定容体积、样品称取、样品重复测定以及标准曲线等产生的不确定度。在检测中应特别注意这几个影响因子,以保障检测结果的准确度。该研究可以反映测量乙基麦芽酚的置信度和准确度,为日常检测提供有利参考。
