利用CRISPR/Cas9 系统构建低DCPC 杂质含量的CPC 工业高产菌种
2021-07-26徐燕,冯涛,储炬
徐 燕, 冯 涛, 储 炬
(1. 华东理工大学生物反应器工程国家重点实验室,上海 200237;2. 国药集团威奇达药业有限公司,山西大同 037300)
头孢菌素C(CPC)被Abraham和Newton 两位学者从顶头孢霉(Acremonium chrysogenum)培养液中分离鉴定[1-2],随后,以CPC 为原料的头孢菌素类抗生素逐渐成为世界抗生素市场的重要一员。该类抗生素属于β-内酰胺类抗生素,具有广谱抗菌性,且相较于青霉素,其疗效更好,安全性更高,副作用更少。此外,由于CPC 中二氢噻嗪环与β-内酰胺环相连的特殊母核结构,使其具有更好的青霉素酶耐受性[3]。
CPC 去除支链后得到的7-氨基头孢烷酸(7-ACA)是各类头孢菌素衍生药物的起点化合物,常通过发酵手段获得。在发酵过程中虽常含有青霉素N(PEN)、脱乙酰氧头孢菌素C(DAOC)等中间产物,但脱乙酰头孢菌素C(DCPC)为主要副产物。DCPC 结构与性质和CPC极为相似,两者均为氨基酸,它常以内铵盐形式存在,发酵生产中DCPC 的质量分数约为CPC 质量分数的15%~20%[4]。在现有的生产工艺中,DCPC 虽能够与主产物CPC 进行分离,但其在余液中的大量残留会造成环境污染。抗生素的长期累积,会使周围土壤微生物与动植物生长进化均受到影响,相关联的上下游生物也会受到干扰,从而影响该地区的生态平衡。虽然目前有许多学者对CPC 废液中副产物的再利用进行了大量的研究,但是由于可操作性与经济效益等问题,真正应用于实际生产的还少之又少。
DCPC 的累积有两方面原因:一方面,顶头孢霉自身DCPC 乙酰转移酶(DCPC-AT)启动子强度不足,转录量低,使DCPC 无法有效转化为CPC,成为生产过程中的限制性步骤之一[5];另一方面,顶头孢霉Ⅷ染色体上含有CPC 乙酰水解酶基因(cahB),菌体经72 h 培养后可编码表达一个1.4 kb 的单链转录子,翻译出的乙酰水解酶(CPC-AH)可将已有的CPC 水解为DCPC,CPC-AH 酶活性虽然从120 h 开始下降,但144 h 仍然能观察到活性[6]。
近年来,原为细菌和古细菌的获得性自我免疫预防机制CRISPR/Cas9 系统[7]在基因编辑领域大放异彩,在酿酒酵母(Saccharomyces cerevisiae)中,已实现多基因[8-9]、多位点[10]、长片段(大于30 kb)[11]的成功编辑,在米曲霉(Aspergillus oryzae)[12]、构巢曲霉(Aspergillus nidulans)[13]、里 氏 木 霉(Trichoderma reesei)[14]等真菌中有多种应用,极大提高了真菌中基因编辑效率。顶头孢霉属于半知菌门真菌,与其他丝状真菌相比,其生长周期长且无有性生殖阶段。本实验室首先在顶头孢霉野生型CGMCC3.3795中构建了CRISPR/Cas9 系统,并对聚酮合酶相关基因(sorA、sorB)分别进行了敲除,编辑效率远高于传统方法[15]。
本研究利用CRISPR/Cas9 系统,对顶头孢霉发酵过程中杂质DCPC 累积问题进行了定向改造,构建基因cahB缺陷菌株与基因cefG过表达菌株,以期降低DPCP 杂质含量,提高CPC 产量与质量。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒 顶头孢霉(Acremonium chrysogenum)1-D1 工业菌株、大肠杆菌(Escherichia coli)DH5α均由本实验室保藏。真菌表达质粒pAN7-1 由本实验室保存,含RGR(Ribozyme-gRNA-Ribozyme)结构质粒pUC57 由华大基因合成,含CRISPRCas9 基因编辑系统质粒pAN7-sorA 由本实验室构建与保存。
1.1.2 培养基与培养方法 麦芽汁培养基(用于工业顶头孢霉菌株斜面培养)、种子培养基(用于顶头孢霉菌株种子培养)、摇瓶发酵培养基(用于顶头孢霉菌株发酵),配方与培养方法参见文献[16],并略有调整(种子培养基pH 为7.2)。原生质体培养基与转化方法参见文献[14]。
1.1.3 仪器与检测方法 利用高效液相色谱仪(日本岛津有限公司LC-20T)对CPC 和DCPC 进行检测。CPC 检测方法参考文献[16]。DCPC 检测:流动相为体积分数50%的甲醇与磷酸盐的混合溶液(体积比为3∶97),流速为1 mL/min,等度洗脱,检测波长为254 nm,柱温为30 ℃,进样量为20 μL,检测时间35 min。
1.1.4 酶与试剂 质粒提取试剂盒、胶回收试剂盒:Axygen 公司;真菌基因组抽提试剂盒、真菌RNA 提取试剂盒:生工生物工程(上海)股份有限公司;Evo M-MLV Reverse Transcriptase 反转录试剂盒与SYBR® Green Premix qPCR 试剂盒:湖南艾科瑞生物工程有限公司;限制性内切酶:NEB 公司;一步克隆试剂盒、Phanta酶等PCR 酶:Vazyme 公司;CPC 标准品与DCPC 标准品(含量未知)由国药集团山西威奇达药业有限公司馈赠。四丁基氢氧化铵(Tetrabutylammonium Hydroxide):色谱级,上海阿拉丁生化科技股份有限公司。流动相磷酸盐(用于CPC 与DCPC 检测):850 mL超纯水中加入0.8 g NaH2PO4,搅拌混匀后,用四丁基氢氧化铵调节pH 至7.3,0.45 μm 滤膜抽滤除杂,超声30 min 去除气泡。
1.1.5 引物合成与测序 测序、引物合成均由生工生物工程(上海)股份有限公司完成。表1 为引物列表,其中大写字母为模板质粒pUC57 中RGR 特异性结构的骨架。

表1 引物列表Table 1 List of primers

续表1
1.2 sgRNA 设计与载体构建
经NCBI 数据库查询,得到顶头孢霉野生型( ATCC11550) CPC 乙 酰 水 解 酶( CPC-AH) 基 因cahB(ACRE_083500),通过与工业菌1-D1 全基因测序结果进行比对,找到工业菌中相对应的基因序列(GME1727_g)。通过sgRNA 设计网站(https://gt-scan.csiro.au/submit/)设计靶目标序列5’-gcggcaagaccggccacatgAGG-3 ’ 。 设 计 引 物 sgcahB-1、 sgcahB-2、sgcahB-3、sgcahB-4(见表1)。以pUC57 为模板,进行PCR 扩增,回收得到长度分别为84 bp 与204 bp的两片段,将片段融合PCR 后获得可识别cahB基因的特异性sgRNA 片段cahB-sgRNA。
将质粒pAN7-sorA 双酶切(HindIII、SpeI)胶回收含Cas9 蛋白表达盒的大片段质粒骨架,与cahBsgRNA 片段一步克隆连接,构建pAN7-cahB质粒。将质粒转大肠DH5α感受态,挑取单菌落,液体培养后提质粒送测序。载体pAN7-cahB 构建电泳图见图1 所示。
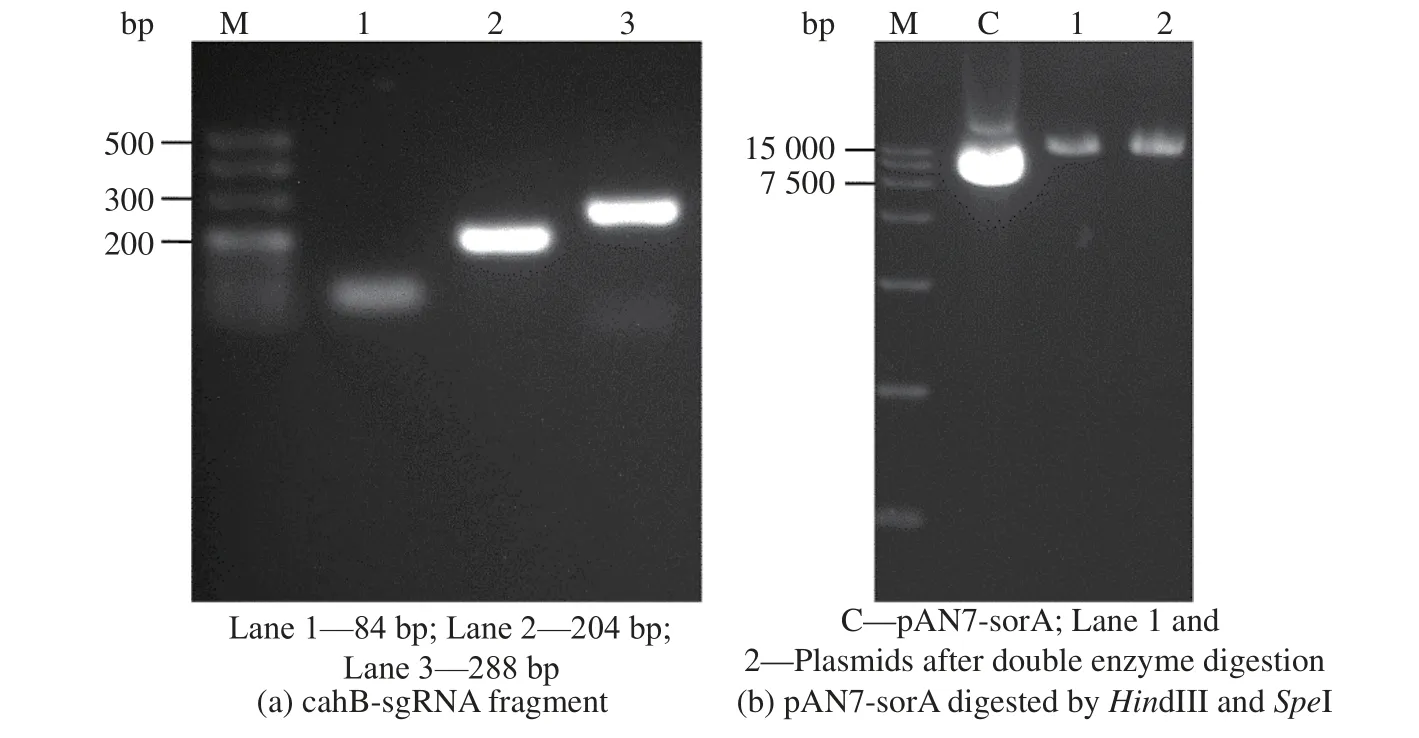
图1 pAN7-cahB 构建电泳图Fig. 1 Electrophoretogram of pAN7-cahB construction
1.3 cefG 基因表达盒与Donor DNA 构建
依据NCBI 数据库中cefG序列(GI: 672 794 352),比对本实验室已有测序结果,获得工业菌株1-D1中cefG序列,设计引物,以1-D1 基因组为模板扩增,获得cefG基因片段,见图2。由图可见,以pAN7-1为模板,设计反向引物(R-pAN7-1-F,R-pAN7-1-R),PCR 得到含启动子与终止子的反向骨架PgpdApAN7-Ttrpc,采用一步克隆的方法将骨架与cefG基因片段连接,构建cefG表达质粒pAN7-1-cefG,进行大肠杆菌感受态转化,挑单菌落PCR 初步验证后,提质粒测序。

图2 pAN7-1-cefG 构建电泳图Fig. 2 Electrophoretogram of pAN7-1-cefG construction
Donor DNA 构建流程与原理见图3。首先,依据1-D1 测序结果,选取cahB基因上下游长度为1 000 bp左右片段作为同源臂,PCR 扩增获得同源臂B1、B2。以Split-Marker 重组技术为原理,设计引物(cahB-PT-F1,cahB-PT-R1)、(cahB-PT-F2,cahB-PTR2),将同源修复片段分上下游片段进行导入。将质粒上PgpdA-cefG-Ttrpc 表达盒分两部分进行扩增得到PgpdA-cefG(2 286 bp)、cefG-TtrpC(2 370 bp),中间含200 bp重叠区。电泳条带见图4(a)。采取融合PCR 方式与上下游同源臂分别连接,获得Donor DNA(B1-PgpdA-cefG、cefG-TtrpC-B2),见图4(b)。
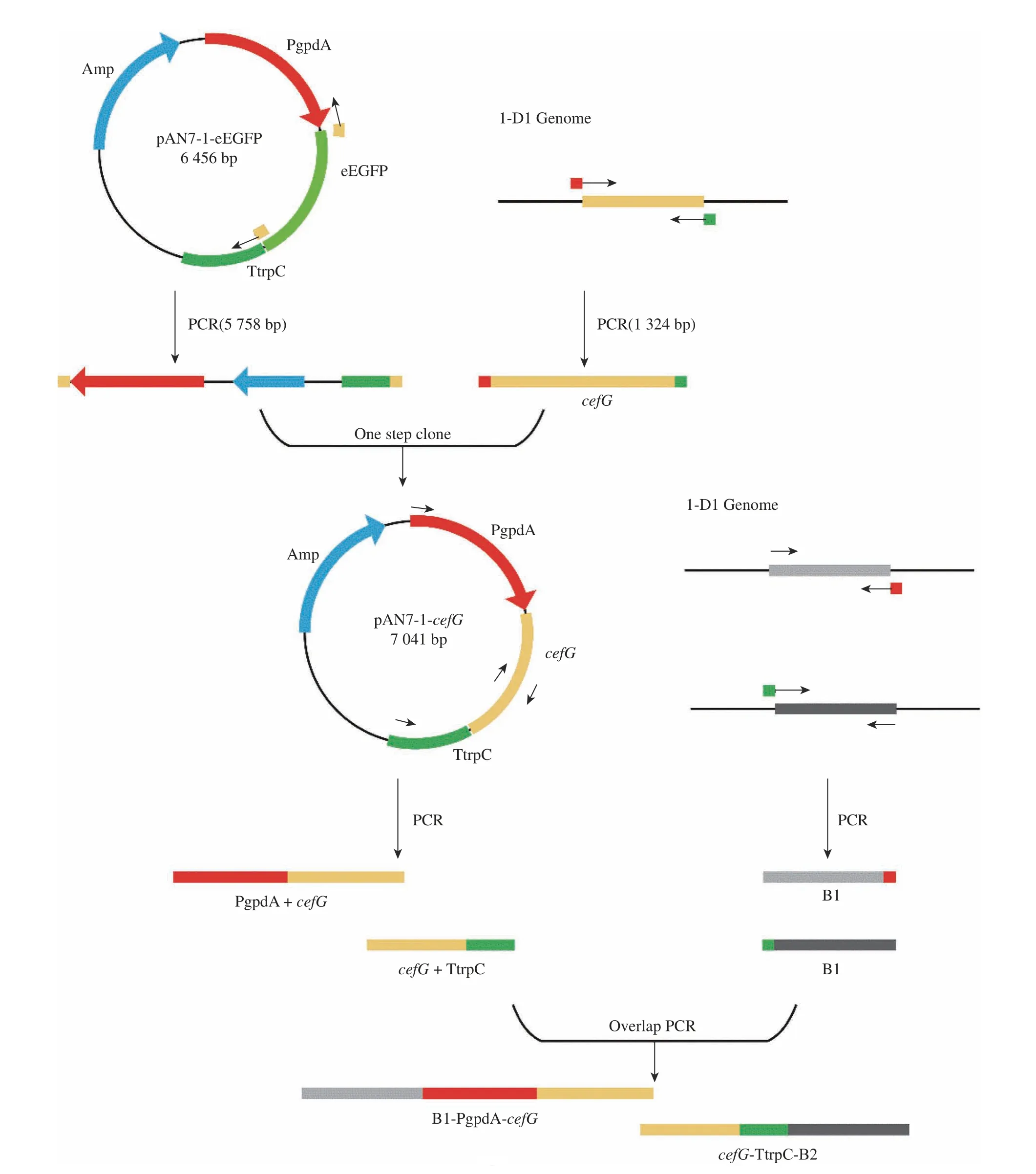
图3 Donor DNA 构建流程图Fig. 3 Donor DNA generation flow chart

图4 Donor DNA 构建电泳图Fig. 4 Electrophoretogram of donor DNA construction
1.4 原生质体转化
将构建完成的pAN7-cahB 质粒与同源修复片段(B1-PgpdA-cefG、cefG-TtrpC-B2)进行顶头孢霉原生质体共同转化,在PEG 介导下,将外源质粒与同源修复表达盒导入菌体内,进行特异性打靶与同源修复。转化时,外源质粒的质量为8~10 μg,pAN7-cahB 质粒、同源修复片段(B1-PgpdA-cefG)和同源修复片段(cefGTtrpC-B2)质量比为4∶1∶1,总体积不超过20 μL(当无需同源修复片段协同作用时,外源质粒所需量不变但总体积不超过10 μL)。当外源核酸物质与原生质体轻柔混匀后,将该体系与上层培养基混合后倒至含有下层转化培养基的平板中,28 ℃培养10~12 d,待转化板长出半透明的白色菌落,使用已灭菌牙签转移至麦芽汁平板(潮霉素抗性:150 μg/mL),28 ℃培养7 d。转化步骤见图5。

图5 原生质体转化实验流程图Fig. 5 Flow chart of protoplast transformation experiment
1.5 转化子筛选验证
1.5.1 Ac-ΔcahB 验证方法 在PAM 序列上下游500~1 000 bp 处设计引物(C-veri-F,C-veri-R),以基因组为模板进行PCR,获得的片段(1 500 bp)通过T7EI 酶切来初步验证是否与基因组有错配碱基,再进行测序验证,结果见图6。转化子测序结果为5’-gcggcaagaccggccac--gAGG-3’,将其与原基因组进行比对,发现PAM 序列(AGG)的上游有2 个碱基缺失。将转化子进行遗传稳定性验证,发现传代至第5 代基因组时碱基缺失仍然存在,说明采用单质粒转化方法进行敲除的菌株,其遗传稳定性良好,没有产生回复突变。
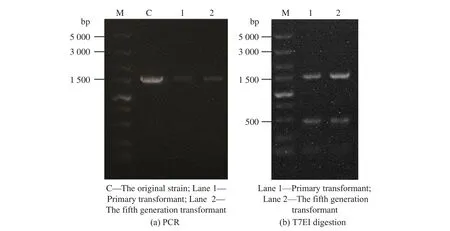
图6 Ac-ΔcahB 转化子验证结果Fig. 6 Ac-ΔcahB transformant verification results
单质粒转化在验证时发现,部分转化子无法进行PCR 获得验证片段,这可能是由于Cas9 蛋白切割造成的双链断裂所引发的非同源性末端接合(NHEJ),导致基因组其他部分的大片段插入与缺失,使验证引物无法与基因组匹配。但是由于其插入与缺失长度的不确定性,无法很好通过调整引物与片段长度解决,故采取单质粒与Donor DNA 共转方式以较好解决突变株验证问题。
1.5.2 Ac-ΔcahB:cefG 验证方法 转化子验证原理见图7。首先,以C1-1、C1-2 为引物扩增靶基因序列,对照组1-D1 有扩增片段,实验组应无扩增片段;而以C2-1、C2-2 为引物扩增同源修复片段中cefG基因表达盒的部分序列,对照组1-D1 应无扩增片段,实验组应有扩增片段;再以C3-1、C3-2、C4-1、C4-2 为引物分别扩增上游同源臂B1 与表达盒启动子部分、表达盒终止子与下游同源臂B2 部分,对照组1-D1应无扩增片段,而实验组应有扩增片段。最后用C3-1、C4-2为引物扩增时,对照组与实验组条带长度有明显差异,电泳结果见图8。
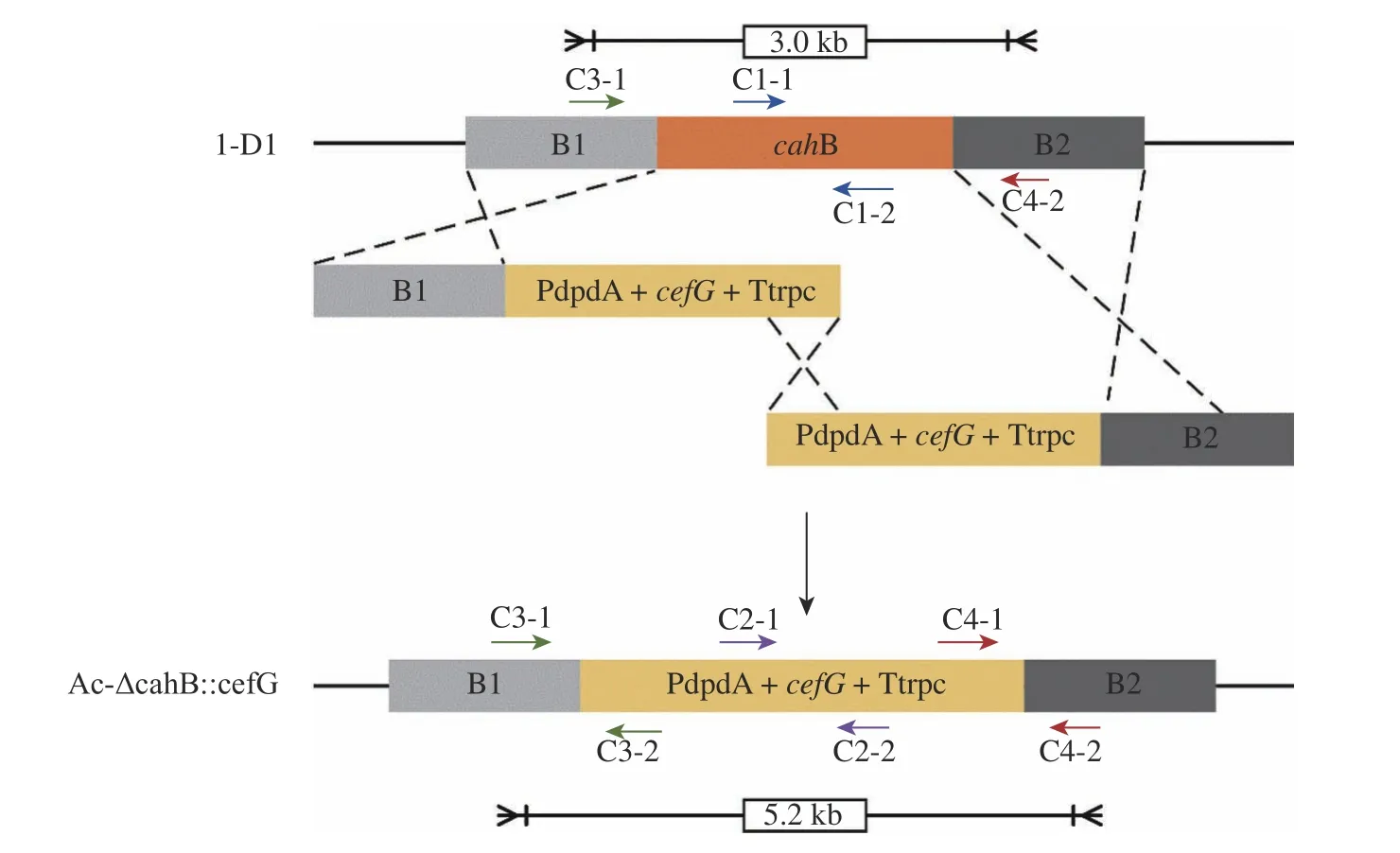
图7 Ac-ΔcahB::cefG 验证原理图Fig. 7 Ac-ΔcahB::cefG verification principle diagram
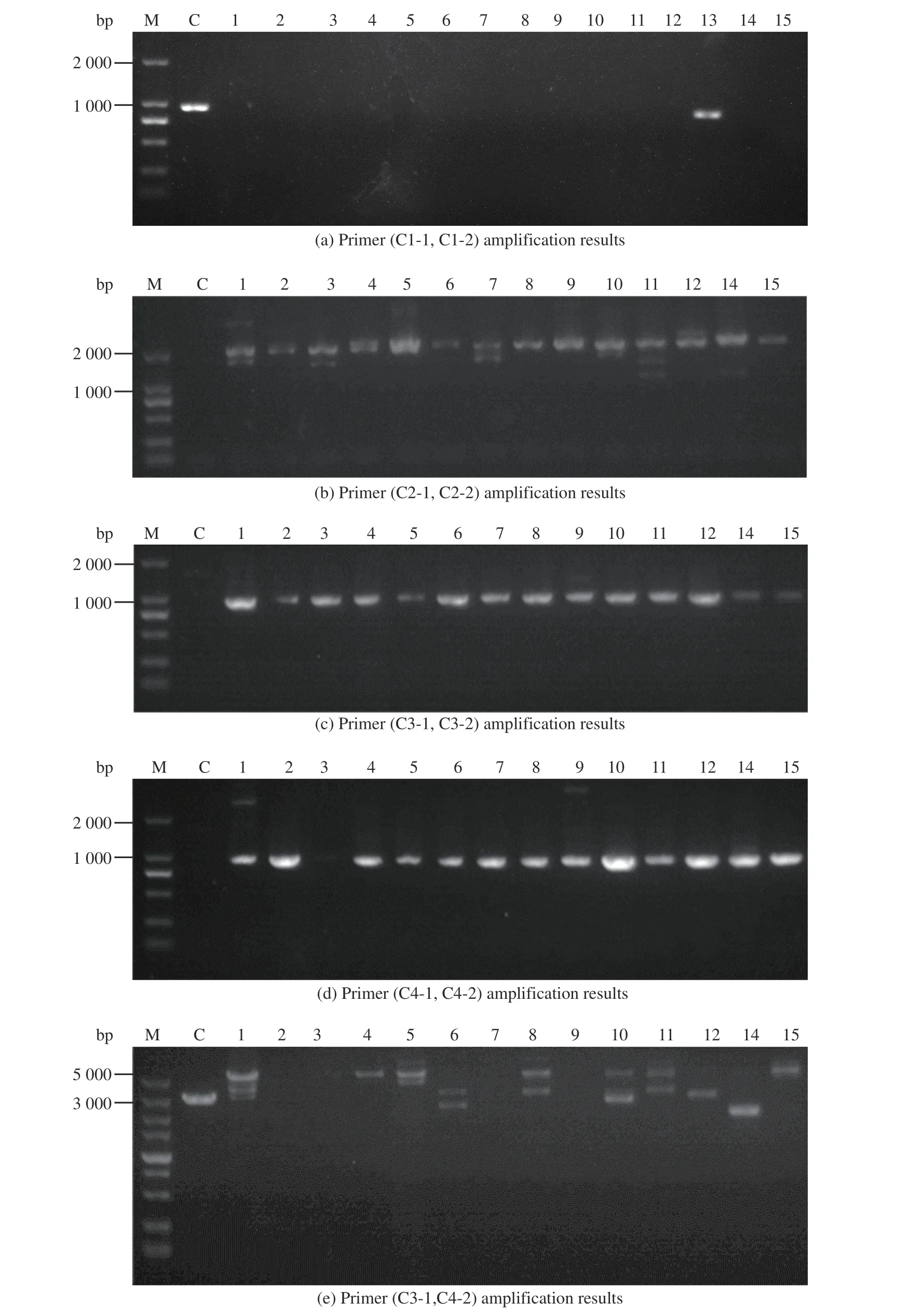
图8 Ac-ΔcahB::cefG PCR 验证图Fig. 8 Ac-ΔcahB::cefG PCR verification chart
1.6 RNA 提取以及RT-PCR(Red Time Quantitative PCR)分析相关基因转录水平
取培养72、96 h 后的顶头孢霉改造菌株Ac-ΔcahB、Ac-ΔcahB::cefG 与出发菌株1-D1 的发酵液,去尽上清并用DEPC(焦碳酸二乙酯)水洗涤3 次后,进行液氮研磨,按照真菌RNA 提取试剂盒说明书要求进行RNA 提取。确认RNA 质量与浓度后,使用Evo M-MLV Reverse Transcriptase 反转录试剂盒说明书进行反转录制备cDNA。以此为模版,以cefG-F1、cefG-R1 为引物,使用SYBR® Green Premix qPCR 试剂盒进行RT-PCR 扩增。
2 结果与讨论
2.1 出发菌株与突变菌株的CPC 产量比较
将突变株Ac-ΔcahB、Ac-ΔcahB::cefG 与对照工业菌株1-D1 进行168 h 的液体摇瓶发酵,对CPC产量与杂质DCPC 产量进行比较研究。CPC 产量如图9所示,突变株Ac-ΔcahB的CPC产量为4 665 μg/mL,出发菌株的CPC 产量为4 578 μg/mL,两者发酵水平相接近。而突变株Ac-ΔcahB::cefG 的CPC 产量为6 072 μg/mL,相较于出发菌株,总体提高了32.6%,差异显著(p=0.025 9)。以上结果可推测,插入强化表达的cefG基因可使DCPC 得到有效转化,减少了菌体中的积累,在降低杂质的同时进一步提高了CPC 的产量。

图9 出发菌株与突变株CPC 产量比较Fig. 9 CPC production comparison of starting strain and mutant strain
cahB基因是CPC 乙酰水解酶基因,可将CPC 水解成为无经济价值的DCPC。但是在此结果中发现,敲除该基因后CPC 并未达到预期中明显提高的效果,这可能由于水解的CPC 在总产量中所占比重比较小。有学者对CPC 乙酰水解酶进行了酶动力学反应研究,结果表明,纯化至均质的酶蛋白与CPC 的亲和力不高(Km=33.7 mmol/L),同时其酶活性在发酵至120 h 时开始逐渐下降[6]。底物CPC 浓度较低时,与乙酰水解酶结合转化的效率较低,水解能力较弱,因乙酰水解酶减少的CPC 在总产量中所占比重比较小,故敲除后CPC 产量没有明显提升。此外,在对基因组进行分析时发现,菌体中有可能存在其他CPC 水解酶,如GME4736_g 与GME1825_g(图10),其分别注释为碱性蛋白酶(Alkaline proteinase)与角质层降解蛋白酶(Cuticle-degrading protease),均具有丝氨酸型肽链内切酶活性,而cahB与枯草芽孢杆菌的丝氨酸蛋白质家族基因具有高同源性。相较于cahB,这些注释为丝氨酸家族的蛋白质基因对CPC 的水解可能起主导作用。
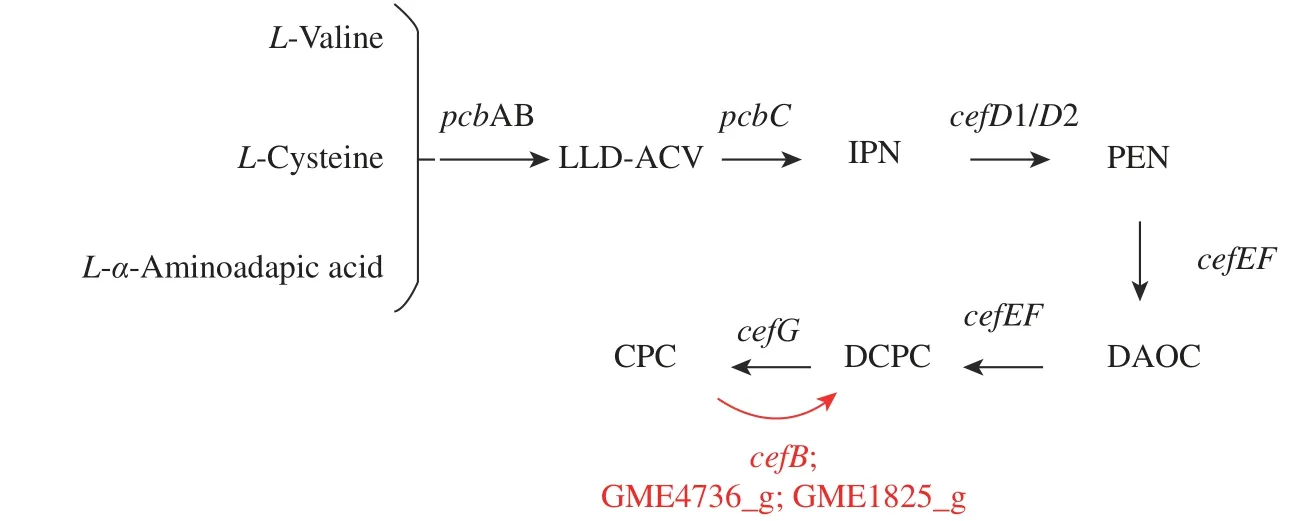
图10 CPC 合成与分解过程的相关基因Fig. 10 Genes involved in the process of CPC synthesis and decomposition
2.2 出发菌株和突变菌株的DCPC 产量比较


图11 出发菌株与突变株发酵液高效液相色谱图Fig. 11 HPLC of the fermentation broth of starting strain and mutant strain

图12 出发菌株与突变株DCPC 的含量比较Fig. 12 DCPC content comparison of starting strain and mutant strain

2.3 基因转录水平测定
以出发菌株1-D1 为对照,提取发酵72 h 与96 h的菌体RNA 进行荧光定量PCR(RT-qPCR)分析。分别以菌株中γ-actin 为内参基因进行矫正,对DCPC乙酰转移酶基因cefG进行表达量测定,结果见图13所示。在顶头孢霉中,DCPC 乙酰转移酶基因cefG内源性启动子较弱,基因表达强度较低一直是CPC 生产过程中的限制性步骤。在Ac-ΔcahB::cefG菌株中,强启动子PgpdA提高了cefG表达量,且上调倍数高达5 倍。DCPC 乙酰转移酶基因转录量的提升可能增加了菌体内乙酰转移酶的含量,从而提高了菌体对中间产物DCPC 的转化效率,促进了CPC 合成。

图13 DCPC 乙酰转移酶基因cefG 相对表达量测定Fig. 13 Determination of cefG expression level of DCPC acetyltransferase gene
3 讨 论
头孢菌素类抗生素因其毒性低、抗菌谱广、作用效果好等特点被广泛应用于临床治疗中,而CPC 作为头孢菌素类抗生素的重要原料,其生产菌株顶头孢霉的发酵却常有中间产物与类似物作为杂质残留,对CPC 产量以及分离纯化造成影响。
本文运用CRISPR/Cas9 基因编辑系统构建了Ac-ΔcahB 菌株,并通过结合Donor DNA,优化了敲除效率,构建了Ac-ΔcahB::cefG 菌株。cahB基因的敲除虽不能有效提高CPC 产量,但是从适宜种龄以及平板培养等其他生长状况而言,该基因的敲除对菌体生长并没有过多影响。或许,相较于以往随机插入的过表达方法,将cahB基因作为定点整合位点,是一个便于验证的不错选择。此外,在对基因组进行分析时发现,菌体中有可能存在其他CPC 水解酶,如碱性蛋白酶(Alkaline proteinase)与角质层降解蛋白酶(Cuticle-degrading protease),均具有丝氨酸型肽链内切酶活性,并且cahB与枯草芽孢杆菌的丝氨酸蛋白质家族基因具有高同源性。相较于cahB,可能这些注释为丝氨酸家族的蛋白质基因对CPC 的水解起着主导作用。插入由强启动子PgpdA过表达的cefG基因,使突变菌株Ac-ΔcahB::cefG 在转录水平表达量提高了5 倍,并且在CPC 生产与DCPC 杂质方面较出发菌株均差异显著,既使CPC 产量提高了32.6%,又使DCPC含量控制在6.81%。此外,发酵培养基的有效利用,提高了经济效益,同时由于DCPC 含量的降低减少了废液中抗生素的残留,从而减小了废液排放对环境的影响。
