盐酸萘甲唑啉的合成工艺研究
2021-07-25李明乐李盼欣张志光张勇
李明乐 李盼欣 张志光 张勇

摘 要:为了解决盐酸萘甲唑啉现有合成工艺落后、质量控制标准低等问题,对盐酸萘甲唑啉合成工艺进行了研究。以ICH Q11对化学药品研发与生产的相关规定为指导,设计一条从源头控制杂质产生的合成路线。以1-萘乙酸为起始原料,依次经过酰胺化、酰胺脱水、催化加成环合和成盐等4步反应得到目标化合物,并对各步骤进行工艺参数优化。结果表明,酰胺化反应中,以二氯甲烷为溶剂,n(1-萘乙酸)∶n(二碳酸二叔丁酯)∶n(氨水)= 1∶1.2∶1.2时,收率为88.6%;脱水反应和加成环合反应中,以N,N-二甲基甲酰胺为溶剂,后处理易操作且獲得的中间体纯度高;成盐反应中,n(萘甲唑啉游离碱)∶n(36%盐酸)= 1∶1.48时,收率为90.1%;经工艺参数优化后的4步反应的总收率为50.83%,产品中未检出欧洲药典所列的4种杂质,也没有产生新的杂质。合成工艺反应条件相对温和,后处理简单,可为盐酸萘甲唑啉的工业化生产提供理论基础。
关键词:有机合成化学;盐酸萘甲唑啉;合成工艺;质量源于设计;1-萘乙酸
中图分类号:R971.3 文献标识码:A
doi:10.7535/hbkd.2021yx03008
Study on the synthesis process of naphazoline hydrochloride
LI Mingle1, LI Panxin1, ZHANG Zhiguang1, ZHANG Yong1,2
(1.School of Chemical and Pharmaceutical Engineering,Hebei University of Science and Technology,Shijiazhuang,Hebei 050018,China;
2.State Key Laboratory Breeding Base-Key Laboratory of Molecular Chemistry for Drug of Hebei Province,Shijiazhuang,Hebei 050018,China)
Abstract:In order to solve the problems of out-of-date synthesis process and lag-behind quality control standards of naphazoline hydrochloride,the synthesis process of aimed product was studied.According to the relevant regulation of ICH Q11 on the development and manufacture of drug substances,the modified synthesis route was designed to control the generation of impurities based on the concept of quality by design.Using 1-naphthylacetic acid as the starting material,the target compound was obtained through amidation reaction,amide dehydration reaction,catalytic addition-cyclization reaction and salt formation reaction.The results show as follows:In the amidation reaction,using dichloromethane as the solvent,n(1-naphthylacetic acid)∶n(di-tert-butyl dicarbonate)∶n(ammonia)=1∶1.2∶1.2,the yield is 88.6%;In the dehydration reaction and the addition-cyclization reaction,N,N-dimethylformamide is used as the solvent,the workup procedure is simple and the intermediate purity is high;In the salt formation reaction,n(naphazoline free base)∶n(36% hydrochloric acid)=1∶1.48,the yield is 90.1%.The overall yield of the four-step reaction after optimization of the reaction conditions was 50.83%.The four impurities listed in the European Pharmacopoeia were not detected in the product,and no new impurities were produced.The synthesis process have advantages of mild reaction condition and operational simplicity,so the result provide a theoretical basis for the industrial production of naphazoline hydrochloride.
Keywords:
organic synthesis chemistry;naphazoline hydrochloride;synthesis process;quality by design;1-naphthylacetic acid
盐酸萘甲唑啉(naphazoline hydrochloride),化学名为2-(1-萘甲基)-4,5-二氢-1H-咪唑盐酸盐,结构式如图1所示[1]。它是一种能快速起效的咪唑啉类拟交感神经激动剂,能够收缩眼内或鼻内小动脉血管[2],用于滴眼剂或滴鼻剂等非处方药,治疗眼部充血或鼻塞症状[3-5]。
文献报道的盐酸萘甲唑啉合成方法按起始原料的不同分为2大类,第1大类方法以1-萘乙酸为起始原料,有2种合成方式。其一是1-萘乙酸在盐酸存在下与乙二胺反应得到盐酸萘甲唑啉[6]。这种方法的缺点是,反应需要在240 ℃高温条件下进行,并且以苯作为萃取剂制得粗品,粗品需经大孔树脂柱脱色处理得到目标产物。其二是使用四氧化三铁纳米粒促进1-萘乙酸与乙二胺发生的酰胺化和分子内环合反应,并使用苯共沸蒸馏除去环合过程中产生的水,收率只有30%[7]。第2大类方法以1-萘乙腈为起始原料,目标产物的获得有3种方式。1)1-萘乙腈先与异丙醇或异丁醇在盐酸作用下生成亚胺酯活性中间体,亚胺酯再与乙二胺反应得到目标物[8]。亚胺酯容易水解,反应需要严格控制无水条件。2)在1-萘乙腈与乙二胺发生反应的过程中加入硫化氢或能够原位产生硫化氢的含硫催化剂(包括二硫化碳、硫化钠或硫脲等),可以在相对温和的条件下获得目标产物[9-10]。文献中未提及催化剂的去除方法。3)采用硫代乙酰胺为催化剂,1-萘乙腈可以与乙二胺发生反应得到目标化合物,反应条件相对较温和且产率较高[11]。反应过程中硫代乙酰胺首先与乙二胺反应,原位产生的硫化氢可以催化1-萘乙腈与乙二胺反应生成产物,伴生的2-甲基-2-咪唑啉具有水溶性,可以方便水洗去除。
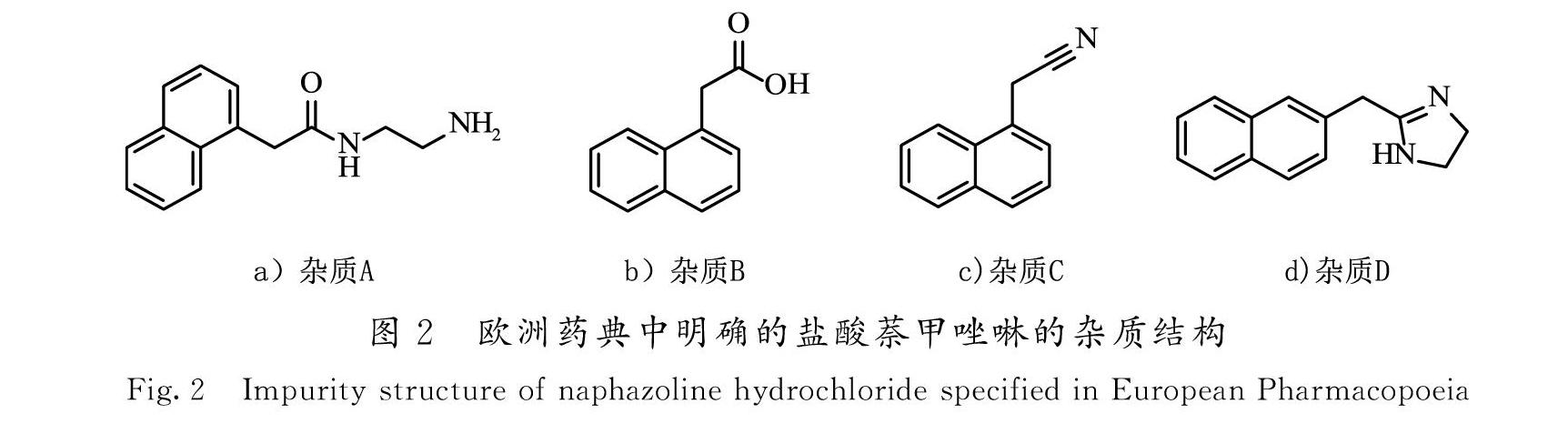
对于盐酸萘甲唑啉原料药,欧洲药典[12]明确了4个杂质结构(如图 2所示)。杂质A为工艺杂质或降解杂质,可归结为分子内环合反应不完全,或由于咪唑啉结构中的亚胺键不稳定开环产生;杂质B是由杂质A继续深度降解产生,也可能是采用第1类方法时未反应的起始原料残留;杂质C是采用第2类方法时未反应的起始原料残留;杂质D是由起始原料的位置异构体衍生而来[13-14]。
为了全面控制杂质对目标产品的关键质量属性(CQA)的影响,结合对药典中涉及的杂质的来源进行分析,可以从2方面入手进行源头控制。一方面,采用更加有优势的咪唑啉成环方法,控制环合后开环反应的趋势,从而控制杂质A,B和C的产生;另一方面,考虑到物料特性或操作条件的改变,越靠近生产工艺的初始阶段,则影响原料药质量的可能性就越小[15],可以通过延长合成步骤,使产生杂质D的前体在多步骤的后处理过程中予以清除。
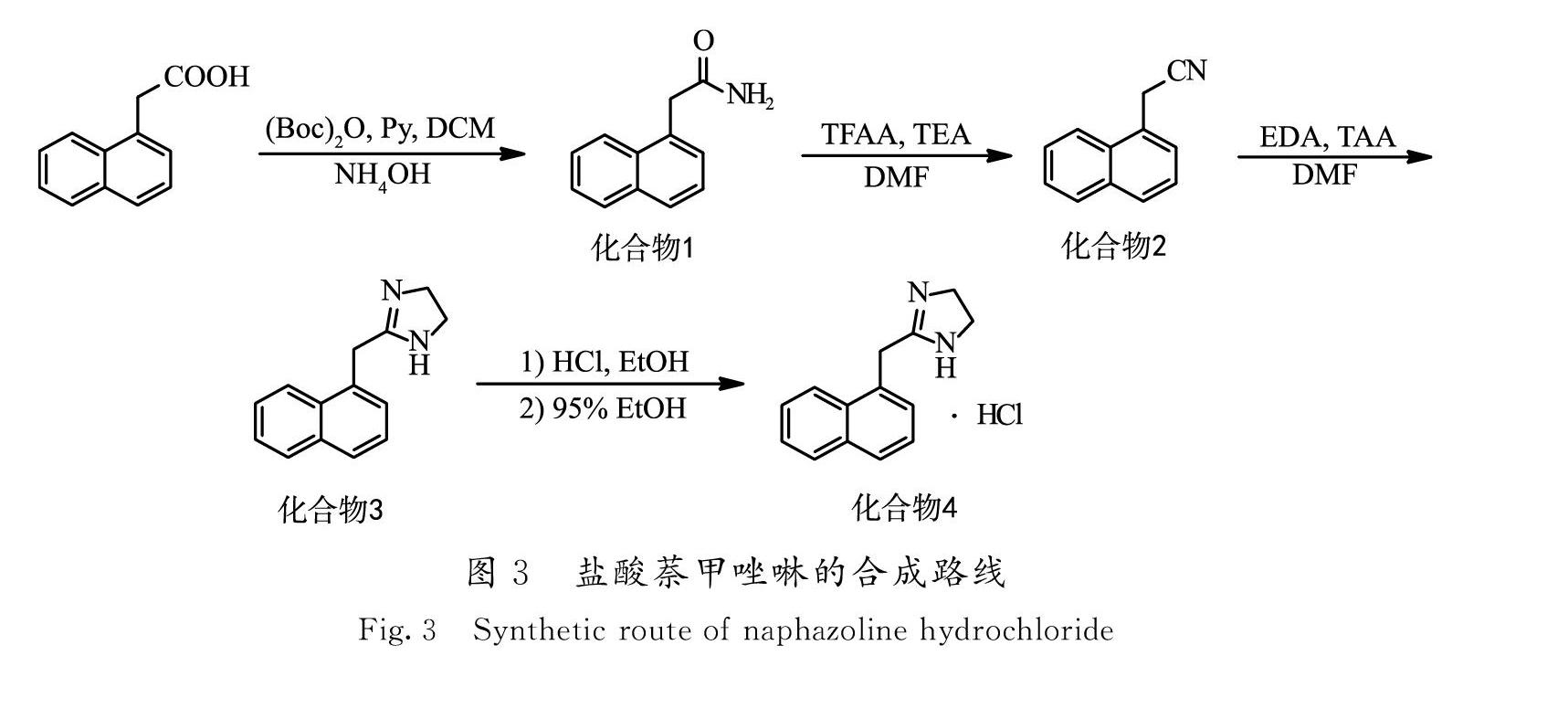
综上所述,设计以1-萘乙酸为起始原料,依次经过酰胺化、酰胺脱水、催化加成环合和成盐等4步反应得到目标化合物。1-萘乙酸是一种植物生长调节剂,作为农药有着广泛的应用,可以视为市售化学品[16-17]。而且通过酰胺化和酰胺脱水2步反应的后处理过程对位置异构杂质的清除作用,降低了杂质D的产生风险。在得到1-萘乙腈后,通过乙二胺与硫代乙酰胺原位生成的硫化氢的催化作用,使加成环合过程更加容易且完全。该路线基于ICH对化学药品研发与生产的相关规定,从反应方法和起始物料的选择2个方面综合考虑,从源头上控制原料药中的杂质,满足药品对产品质量的严格要求,反应路线如图3所示。
1 实验部分
1.1 主要仪器与试剂
Bruck-400型核磁共振仪,瑞士布鲁克公司提供;X-4精密显微熔点测定仪,上海精密科学仪器有限公司提供;ZF-2三用紫外分析仪,上海市安亭电子仪器厂提供;电热鼓风干燥箱,天津市泰斯特仪器有限公司提供;LC/MS-2020液相色谱质谱联用仪,日本岛津公司提供。
1-萘乙酸,二碳酸二叔丁酯,三氟乙酸酐(TFAA),硫代乙酰胺(TAA),乙二胺(EDA),由阿拉丁试剂有限公司提供;其他试剂均由天津市永大化学试剂有限公司提供。
1.2 试验方法
1.2.1 酰胺化反应(化合物1的合成)
将96.82 g(0.52 mol)1-萘乙酸加入到500 mL二氯甲烷中,搅拌使其溶解,然后加入16.61 g(0.21 mol)吡啶和42.43 g(0.624 mol)氨水。控制温度为20 ~ 25 ℃,滴加136.18 g(0.624 mol)二碳酸二叔丁酯,滴毕,室温反应3 ~ 4 h。采用TLC(展开剂:V(石油醚)∶V(乙酸乙酯)∶V(乙酸)= 20∶20∶1)监测反应终点。抽滤,干燥后得到85.33 g白色结晶状固体,收率为88.6%,熔点为180.1 ~ 184.5 ℃。1H-NMR(600 MHz,DMSO)δ 8.08(d,J=8.4 Hz,1H),7.91(d,J=7.8 Hz,1H),7.80(d,J=7.8 Hz,1H),7.58(s,1H,NH2),7.55 ~ 7.42(m,5H),7.01(s,1H,NH2),3.87(s,2H,Ar-CH2)。
1.2.2 酰胺脱水反应(化合物2的合成)
依次将80.01 g(0.432 mol)化合物1,118.39 g(1.17mol)三乙胺加入到125 mL N,N-二甲基甲酰胺中,搅拌使原料充分溶解。冰水浴降温至-10 ℃,滴加113.52 g(0.54 mol)三氟乙酸酐,通过控制滴加速度使反应温度维持在-10~-5 ℃,滴毕,维持该温度反应1 h。TLC(展开剂:V(石油醚)∶V(乙酸乙酯) = 5∶1)监测反应终点。向反应体系中滴加500 mL水,滴毕,在0~10 ℃搅拌30 min。抽滤,得到化合物2的粗品。将粗品用500 mL甲苯溶解,水洗涤(50 mL×3),无水硫酸钠干燥,减压浓缩除去溶剂,放冷固化后得到66.41 g淺黄色固体,收率为91.9%,熔点为33.5~37.2 ℃。1H-NMR(600 MHz,CDCl3)δ 7.83(d,J=8.4 Hz,1H),7.77(d,J=7.8 Hz,1H),7.74(d,J=7.8 Hz,1H),7.53~7.47(m,3H),7.38(m,1H),3.96(d,J=7.2 Hz,2H)。
1.2.3 加成环合反应(化合物3的合成)
将65.04 g(0.389 mol)化合物2和46.76 g(0.778 mol)乙二胺、2.93 g(0.039 mol)硫代乙酰胺依次加入到65 mL N,N-二甲基甲酰胺中,100~105 ℃反应3 h。TLC(展开剂:V(二氯甲烷)∶V(甲醇)=10∶1)监测反应终点。停止加热,待反应体系降温至45 ℃,向反应瓶中缓慢滴加260 mL水,室温搅拌1 h,抽滤,减压干燥,得到白色固体粉末70.76 g,收率为86.5%(文献[10]收率为84.0%),熔点为114.9~120.1 ℃。1H NMR(600 MHz,DMSO)δ 8.15(d,J=8.4 Hz,1H),7.92~7.90 (m,1H),7.81~7.79(m,1H),7.54~7.49(m,2H),7.48~7.42(m,2H),6.31(s,1H,NH),3.89(s,2H,Ar-CH2),3.37(s,4H,2×CH2)。
1.2.4 盐酸萘甲唑啉(化合物4)的合成及精制
将67.30 g(0.32 mol)化合物3加入到280 mL无水乙醇中,搅拌15 min使原料充分溶解,室温下滴加55.34 g(0.47 mol)盐酸。加毕,继续搅拌15 min,抽滤,减压干燥,得到盐酸萘甲唑啉粗品71.14 g,收率为90.1%。
向上步反应得到的70.00 g粗品中加入350 mL 95%乙醇,搅拌加热回流,待固体全部溶解后,加入3.50 g活性炭脱色10 min。抽滤,滤液在-5~0 ℃搅拌1 h,至析晶完全。抽滤,将滤饼用无水乙醇洗涤(20 mL×3),50 ℃真空干燥3 h,得到白色结晶56.07 g,产率为80.1%,纯度为100% [HPLC面积归一化法:色谱柱Waters Symmetry C8 (4.6 mm×250 mm,5 μm);以乙腈为流动相A,以1.57 g/L辛烷磺酸钠溶液 (含0.5%乙酸) 为流动相B,进行梯度洗脱;检测波长为280 nm,柱温30 ℃,流速为1.0 mL/min]。1H-NMR(600 MHz,DMSO)δ 10.41(s,2H,NH·HCl),8.10(d,J=8.4,1H),8.00(m,1H),7.95(d,J=8.4 Hz,1H),7.63~7.58(m,3H),7.55(m,1H),4.41(s,2H,Ar-CH2),3.81(s,4H,2×CH2)。13C NMR(126 MHz,DMSO)δ 169.92,133.88,131.70,129.31,129.22,128.98,128.61,127.36,126.66,126.18,123.82,44.73,29.79。
2 结果与讨论
2.1 酰胺化反应影响因素
羧酸转化为酰胺的常用方法是将羧酸在氯化试剂(如氯化亚砜)的作用下转化为酰氯,然后和氨水反应制得酰胺,但会产生大量的氯化氢和二氧化硫等酸性气体。参考文献[18],使用二碳酸二叔丁酯作为羧酸的活化试剂,将1-萘乙酸转化为化合物5(见图4),与氨水发生氨解反应得到化合物1,化合物1以固体的形式从反应液中析出。
实验中发现,如果按照预想首先使用二碳酸二叔丁酯活化羧酸,然后再滴加氨水进行氨解反应,过程中生成一个不能继续转化的新点,经柱层析分离鉴定为化合物6,推测其是在活化过程中生成的化合物5和另外一分子的1-萘乙酸发生交换反应。得到的化合物6相对稳定,不能被氨水氨解。为了避免化合物6的生成,通过改变加料顺序,氨水先于二碳酸二叔丁酯加入反应体系,1-萘乙酸与二碳酸二叔丁酯反应生成的化合物5可以优先与氨水反应,生成化合物1,反应机理如图4所示。
考查了二碳酸二叔丁酯和氨水配比对收率的影响,如表1所示。从反应成本及产品质量角度考虑,最终选择采用n(1-萘乙酸)∶n(二碳酸二叔丁酯)∶n(氨水)=1∶1.2∶1.2作为配比参数进行反应。
2.2 酰胺脱水反应影响因素
参考文献[19]—文献[21]报道的方法,使用三氟乙酸酐-三乙胺为脱水剂。该反应的影响因素主要包括反应温度、物料配比和后处理方法等。根据文献报道及同类型反应经验,将反应温度控制在-10~-5 ℃,反应即可顺利进行。物料配比对反应收率的影响结果如表2所示。由表2可知,三氟乙酸酐用量少于1.25 eq时,原料反应不完全;三氟乙酸酐用量大于1.25 eq时,反应产生了更多的副产物。因此,三氟乙酸酐的用量选择1.25 eq进行反应为最优条件。选用N,N-二甲基甲酰胺作为溶剂,用量小,且后处理方便。
2.3 加成环合反应影响因素
参考文献[11],将催化量的硫代乙酰胺首先和乙二胺反应生成硫化氢,硫化氢分子可以快速与氰基发生加成反应,生成亚氨基硫代羧酸中间体,该中间体作为桥梁可以和乙二胺在相对温和的条件下反应,得到萘甲唑啉游离碱。试验过程中发现,如果不添加反应溶剂,乙二胺即是反应物又是反应溶剂,后处理困难。选择用N,N-二甲基甲酰胺作为反应溶剂进行反应,反应结束后加水淬灭反应,产物以固体的形式析出,通过过滤可除去反应中产生的水溶性杂质。
反应温度和反应时间对收率的影响结果如表3所示,温度较低时,反应速率慢且反应不完全;温度过高,反应收率无明显变化。因此,反应温度控制在100~105 ℃为最优条件。
2.4 成盐精制反应影响因素
实验过程中发现1.48 eq的盐酸恰好可使化合物3反应完全转化为化合物4。从反应效果及成本考虑,最终选择1.48 eq的盐酸进行成盐反应。另外,重结晶溶剂对反应收率的影响结果如表4所示,发现使用95%乙醇为重结晶溶剂时收率最高。
3 结 语
以盐酸萘甲唑啉原料药产品质量标准中杂质的控制为研究目标,通过对药典中涉及的杂质来源进行分析,从起始原料的选择和反应工艺2方面进行源头控制。起始原料选择方面:以市售化学品1-萘乙酸为起始原料,依次经过酰胺化、酰胺脱水、催化加成环合和成盐等4步反应得到目标化合物,通过多步化学转化和后处理过程,位置异构体杂质得到了有效控制和清除。反应工艺方面:在酰胺化反应中,通过改变加料顺序减少副产物的生成,从而提高中间体纯度;脱水反应以三氟乙酸酐-三乙胺為脱水剂,在相对温和的条件下实现了酰胺到腈的转化;加成环合反应中,通过加入催化量的硫代乙酰胺,原位产生硫化氢,在相对温和的条件下反应得到萘甲唑啉游离碱;成盐精制反应中,n(萘甲唑啉游离碱)∶n(36%盐酸)= 1∶1.48时,可使成盐反应充分,选用95%乙醇为重结晶溶剂,收率质量较为理想。通过优化工艺参数,4步反应总收率达到50.83%。
完成了盐酸萘甲唑啉原料药的实验室规模实验,对于中试以及工业化放大生产,需要对工艺风险评估、技术转移方案的制订和实施等方面进行逐步推进,针对是否存在规模效应、原材料来源、设备、控制参数等变化因素对质量、收率的影响进行深入研究[22],验证其应用价值。
参考文献/References:
[1] ENNA S J,BYLUND D B.xPharm:The comprehensive pharmacology reference-naphazoline[M].Netheland:Elsevier Inc,2007.
[2] SACHER F,TSCHAIKIN M,KOELSCH S.Novel Use for Alpha Sympathomimetics Having a 2-Imidazoline Structure[P].US:2008108685(A1),2008-05-08.
[3] RAMEY J T,BAILEN E,LOCKEY R F.Rhinitis medicamentosa[J].Journal of Investigational Allergology & Clinical Immunology,2006,16(3):148-155.
[4] 刘院斌,刘爱梅.盐酸萘甲唑啉滴眼液治疗儿童春季过敏性结膜炎的临床观察[J].山西医药杂志,2009,38(2):152-153.
[5] 许益飞,施晓琼,朱元奉,等.复方盐酸萘甲唑啉鼻用喷剂治疗慢性单纯性鼻炎的临床观察[J].海军医学杂志,2011(1):22-23.
XU Yifei,SHI Xiaoqiong,ZHU Yuanfeng,et al.Studies on the efficacy and safety of compound naphazoline hydrochloride nasal spray in treating chronic simple rhinitis[J].Journal of Navy Medicine,2011(1):22-23.
[6] 趙哲勋.盐酸萘甲唑啉合成工艺条件及精制方法的改进[J].天津化工,1991,5(4):37-38.
[7] POPOV Y V,MOKHOV V M,KALITINA I I.Colloid and nanosized catalysts in organic synthesis:Ⅻ.Synthesis of 2-R-2-imidazolines catalyzed by copper and iron oxide nanoparticles[J].Russian Journal of General Chemistry,2016,86(2):281-285.
[8] NAKANISHI S C,SAITO T S.Production of Naphazoline or Its Salt[P].Japan:19930166031,1994-12-20.
[9] 黄彩逢,黎万,熊伟梅,等.一种盐酸萘甲唑啉的制备方法[P].中国:201910438890,2019-07-23.
[10]LEVESQUE G,GRESSIER J C,PROUST M.4,5-Dihydroimidazoles from dithiocarboxylic esters,thiocarboxamides,or nitriles[J].Synthesis,1981(12):963-965.
[11]DASH P,KUDAV D P,PARIHAR J A.Thioacetamide catalysed transformation of nitriles to 2-substituted imidazolines[J].Journal of Chemical Research,2004(7):490-491.
[12]Council of Europe.European Pharmacopoeia[M].Stasbourg:European Directorate for the Quality of Medicines & HealthCore (EDQM),2020.
[13]SAYED N,HEGAZY M,ABDELKAWY M,et al.Spectrophotometric,chemometric and chromatographic determination of naphazoline hydrochloride and chlorpheniramine maleate in the presence of naphazoline hydrochloride alkaline degradation product[J].Bulletin of Faculty of Pharmacy,Cairo University,2013,51(1):57-68.
[14]李苗,童颖,乔戈,等.盐酸萘甲唑啉滴鼻液质量评价[J].医药导报,2021,40(3):369-373.
LI Miao,TONG Ying,QIAO Ge,et al.Quality evaluation of naphazoline hydrochloride nasal drops[J].Herald of Medicine,2021,40(3):369-373.
[15]EMA,CHMP,ICH.ICH Q11 Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities)[EB/OL].[2013-02-11].https://www.ema.europa.eu/en/ich-q11-development-manufacture-drug-substances-chemical-entities-biotechnological biological.
[16]丁益,王百年,韩效钊,等.α-萘乙酸的合成方法及应用前景[J].安徽化工,2004(3):17-18.
DING Yi,WANG Bainian,HAN Xiaozhao,et al.The synthesis method of α-naphthylacetic acid and its future[J].Anhui Chemical Industry,2004(3):17-18.
[17]刘建霞,钟文星,王润梅,等.萘乙酸喷施对盐胁迫下黄芪幼苗的缓解作用[J].中药材,2018,41(1):28-32.
[18]PATTERSON D E,POWERS J D,LEBLANC M,et al.Development of a practical large-scale synthesis of denagliptin tosylate[J].Organic Process Research & Development,2009,13(5):900-906.
[19]张勇,张俏艳,刘甜甜.氟吡菌酰胺的合成工艺改进[J].河北科技大学学报,2017,38(3):263-268.
ZHANG Yong,ZHANG Qiaoyan,LIU Tiantian.Improved synthesis process of fluopyram[J].Journal of Hebei University of Science and Technology,2017,38(3):263-268.
[20]门靖,丁志新,曹卫凯,等.2-氰基-4-吡啶羧酸甲酯的合成工艺改进[J].精细化工中间体,2018,48(2):55-58.
MEN Jing,DING Zhixin,CAO Weikai,et al.An improved process for the preparation of 2-cyano-4-pyridinecarboxylic acid methyl ester[J].Fine Chemical Intermediates,2018,48(2):55-58.
[21]AUGERI D J,ROBL J A,BETEBENNER D A,et al.Discovery and preclinical profile of saxagliptin (BMS-477118):A highly potent,long-acting,orally active dipeptidyl peptidase Ⅳ inhibitor for the treatment of type 2 diabetes[J].Journal of Medicinal Chemistry,2005,48(15):5025-5037.
[22]JOHN A,BLACKER A.制藥工艺开发:目前的化学与工程挑战[M].朱维平,译.上海:华东理工大学出版社,2016.
