基于16S rDNA高通量测序分析水牛初乳与常乳中细菌多样性
2021-07-23谢华德唐振华谢耀锋郭艳霞黄钰涵杨承剑
谢 芳,谢华德,唐振华,谢耀锋,郭艳霞,黄钰涵,杨承剑
(中国农业科学院广西水牛研究所,广西水牛遗传繁育重点实验室,广西南宁 530001)
我国水牛资源丰富,广西水牛数量更是位居全国首位。近年来,在政府扶植和政策驱动下,奶水牛产业已经成为广西的特色优势产业[1],水牛奶产量也升至全国之最。水牛奶因其优良的品质和香醇的口感而被世界公认为“奶中珍品”[2],其蛋白质、脂肪、乳糖、维生素等含量均高于黑白花奶[3]与荷斯坦奶牛相比,水牛适应能力强,具有较强的抗病性和免疫力,迄今尚未有水牛疯牛病例报道[4],尤其是水牛初乳,因其含有大量的免疫和生长因子而一度成为市场新宠,更有研究表明水牛初乳中的免疫球蛋白G、溶菌酶、类胰岛素生长因子等是普通牛乳的5~10 倍[5],因此,水牛乳具有较好的食用安全性和较高的商业价值,深加工优势明显。
近年来,随着分子生物学的发展,高通量测序作为“下一代”测序技术已迅速发展,它避开了微生物分离培养的过程,能检测到纯培养和非培养技术不能发现的低丰度细菌种类,相较传统的测序技术,具有准确率高、耗时短以及信息处理量大等优点[6]。目前,高通量测序技术现已应用于各个领域,包括海洋、土壤、植物和肠道环境,而利用高通量测序对乳品领域进行研究的报道较少[7],以往通过传统测序可以检测到可培养和含量较高的菌落,但很难从样品中获得曾经存活或难以分离的微生物,进而不能全面了解微生物的多样性[8]。牛乳是微生物最好的培养基,这些微生物可能来自患乳房炎奶牛的乳房、挤奶工具亦或是牛棚环境[9],而生奶中致病细菌以及腐败细菌的分布可反映奶水牛的健康状况、水牛奶的污染情况等[10]。作为生产乳品的基础,其微生物含量则是衡量水牛乳品质的关键指标,故了解水牛原料乳中微生物多样性至关重要。
本研究拟利用16S rDNA高通量测序技术,对广西水牛研究所种畜场存栏量较大的三品杂水牛(摩拉×尼里-拉菲×本地沼泽型)初乳和常乳组样本中的细菌群落结构进行比较分析,以期全面了解水牛初乳和常乳中细菌群落结构和多样性差异,旨在为后续水牛乳加工、安全评估等提供参考。
1 材料与方法
1.1 材料与仪器
生水牛乳样本 均取自广西水牛研究所水牛种畜场;E.Z.N.A.® Soil DNA Kit DNA抽提试剂盒,美 国Omega Bio-Tek公 司;biowest agArose琼 脂糖 西班牙Biowest;NEXTFLEX ® Rapid DNASeq Kit建库试剂盒 美国Bioo Scientific;MiSeq Reagent Kit v3 测序试剂盒 美国Illumina;磷酸缓冲盐溶液 赛默飞世尔科技;蔗糖缓冲液 北京雷根生物;溶菌酶、变溶菌素 上海联硕宝为生物;蛋白酶K、RNase、PE Buffer上海生工生物;氯化钠、冰异丙醇、苯酚、氯仿、异戊醇 上海北诺生物科技;糖原 北京百奥莱博。
Eppendorf 5430 R离心机、Eppendorf 5424R高速台式冷冻离心机 德国Eppendorf;NanoDrop2000超微量分光光度计 美国Thermo Fisher Scientific;BioTek ELx800 酶标仪美国Biotek;Quantus™Fluorometer微型荧光计 美国Promega公司;DYY-6C电泳仪 北京市六一仪器厂;ABI GeneAmp®9700 型PCR仪 美国ABI;Illumina Miseq测序仪 美国Illumina。
1.2 实验方法
1.2.1 样本采集 生水牛乳样本随机选取9 头处于围产期的三品种高代杂交水牛(摩拉×尼里-拉菲×本地沼泽型水牛),于每头牛产犊后的第1 d开始取样,连续采集3 d,每头牛采集2 管平行,将每头牛采集到的6 管奶样混匀为混合奶样,定义为初乳(CR4),共9 个样本,编号为C1、C2、C3、C4、C5、S2、S3、S4、S5。于每头牛产犊后的第7 d开始采集奶样,连续3 d,每头采集2 管平行,同样将6 管奶样混匀成混合奶样,定义为常乳(PR),共9 个样本,编号为M1、M2、M3、M4、M5、M6、M7、M8、M9。采样前用温水清洗乳头,人工佩戴无菌手套挤奶,去掉前数把奶后再留样,样本经50 mL无菌PC离心管密封好,置于采样冷藏保温箱迅速带回实验室,于30 min内进行生乳中细菌总脱氧核糖核酸(DNA)的粗提取。
1.2.2 总DNA粗提和PCR扩增 取40 mL生水牛乳样本,添加8 mL乙二胺四乙酸二钠(EDTA)(500 mmol/L,pH8.0),在6500 r/min下4 ℃冷冻离心30 min,用灭菌脱脂棉去掉乳脂层,弃上清液,加入磷酸缓冲盐溶液(PBS)至10 mL,12000 r/min高速冷冻离心10 min,用1 mL蔗糖缓冲液冲洗两次(每次12000 r/min,离心10 min),后悬浮400 μL蔗糖缓冲液及2U变溶菌素和800 μg溶菌酶,37 ℃培养1 h;取上述培养后的样液,分别加入5 μL SDS,12 μL EDTA,5 μL蛋白酶K及78 μL蔗糖缓冲液,55 ℃孵育1 h,再加入65 μL蔗糖缓冲液和135 μL氯化钠)(5 mmol/L),最后加入2 μL RNase于37 ℃孵育30 min,用700 μL苯酚:氯仿:异戊醇(25:24:1)消除蛋白,上下摇匀10 次以上,然后18000×g、4 ℃离心15 min提取上清,重复3 次,最后加入700 μL冰异丙醇(−20 ℃预冷)和20 μg糖原(glycogen,Roche Diagnostic),混匀放置5 min,然后-20 ℃沉淀过夜。取出过夜样本,18000×g离心30 min获取沉淀,用70%冰乙醇清洗两遍(500~750 μL,弹起白色沉淀,13000 r/min离心10~15 min),倾倒上清,将离心管倒放,风干DNA,加入50 μL PE Buffer,70 ℃孵育5 min,用NanoDrop2000 测定DNA浓度。
用NanoDrop2000 检测DNA纯度和浓度,为避免序列太长影响测序,同时兼顾扩增特异性,选用338F(5’-ACTCCTACGGGAGGCAGCAG-3’)和806R(5’-GGACTACHVGGGTWTCTAAT-3’)对16S rDNA基因V3-V4 可变区进行PCR扩增,反应条件如下:95 ℃预变性3 min,27 个循环(95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸30 s),然后72 ℃稳定延伸10 min,最后在4 ℃进行保存。聚合酶链式反应(PCR)反应体系为:5×TransStart FastPfu缓冲液4 μL,2.5 mmol/L dNTPs 2 μL,上游引物(5 μmol/L)0.8 μL,下游引物(5 μmol/L)0.8 μL,TransStart FastPfu DNA聚合酶0.4 μL,模板DNA 10 ng,补足至20 μL。每个样本3 个PCR重复,将3 个重复的PCR产物混合,使用2%琼脂糖凝胶电泳检测产物。
1.2.3 Illumina Miseq测序 使用2%琼脂糖凝胶回收PCR产物,用AxyPrep DNA Gel Extraction Kit进行纯化,Tris-HCl洗脱,2%琼脂糖电泳检测。利用QuantiFluor™-ST进行检测定量。根据Illumina MiSeq平台标准操作规程,将纯化后的扩增片段构建PE 2*300 文库。文库构建步骤:a.接“Y”字形接头;b.使用磁珠筛选去除接头自连片段;c.利用PCR扩增进行文库模板的富集;d.磁珠回收PCR产物得到最终的文库。利用Illumina公司的Miseq PE300 平台进行测序(上海美吉生物医药科技有限公司)。原始数据上传至美国国家生物信息中心(NCBI)数据库进行比对分析。
1.2.4 细菌组成与丰度差异分析
1.2.4.1 稀释曲线分析 构建方法是统计每个样本中,每个OTU所含的序列数,将OTUs按丰度(所含有的序列条数)由大到小等级排序,再以OTU的排序等级为横坐标,以每个OTU中所含的序列数(也可用OTU中序列数的相对百分含量)为纵坐标作图。
1.2.4.2 OUT、韦恩图分析 选用相似水平为97%的OTU对序列进行分类操作单元(OUT)聚类。软件:采用R语言工具统计和作图。
1.2.4.3 群落柱形图(Bar图)直观呈现各样本在(如域、界、门、纲、目、科、属、种、OTU等)分类学水平上含有哪些优势物种以及样本中各优势物种的相对丰度(所占比重)。软件:基于tax_summary_a文件夹中的数据表,利用R语言工具作图。
1.2.5 细菌多样性比较分析
1.2.5.1 Heatmap图 根据物种或样本间丰度的相似性进行聚类,并将结果呈现在群落Heatmap图上,可使高丰度和低丰度的物种分块聚集,通过颜色变化来反映不同分组(或样本)在各分类学水平上群落组成的相似性和差异性。软件及算法:R语言vegan包。
1.2.5.2 主成分分析(PCA分析)通过分析不同样本群落组成可以反映样本间的差异和距离,PCA运用方差分解,将多组数据的差异反映在二维坐标图上,坐标轴取能够最大反映样品间差异的两个特征值。如样本物种组成越相似,反映在PCA图中的距离越近。软件:R语言PCA统计分析和作图。
1.2.5.3 偏最小二乘法判别分析(PLS-DA)是多变量数据分析技术中的判别分析法。通过对主成分适当的旋转,PLS-DA可以有效地对组间观察值进行区分,并且能够找到导致组间区别的影响变量。这种分析方法有利于发现组间的异同点。软件:R语言mixOmics包中plsda分析和作图。
1.3 数据处理
使用Trimmomatic软件原始测序序列进行质控,使用FLASH软件进行拼接:过滤reads尾部质量值20 以下的碱基,设置50 bp的窗口,如果窗口内的平均质量值低于20,从窗口开始截去后端碱基,过滤质控后50 bp以下的reads,去除含N碱基的reads;根据PE reads之间的overlap关系,将成对reads拼接(merge)成一条序列,最小overlap长度为10 bp;拼接序列的overlap区允许的最大错配比率为0.2,筛选不符合序列;根据序列首尾两端的barcode和引物区分样本,并调整序列方向,barcode允许的错配数为0,最大引物错配数为2。使用的 UPARSE软件(version 7.1 http://drive5.com/uparse/),根据97%的相似度对序列进行分类操作单元(OUT)聚类并剔除嵌合体。利用RDP classifier(http://rdp.cme.msu.edu/)对每条序列进行物种分类注释,比对 Silva数据库(SSU128),设置比对阈值为70%。
菌群多样性分析及绘图:均采用R语言进行统计分析和作图[11]。
2 结果与分析
2.1 测序数据、OTU及稀释曲线分析
通过对初乳和常乳组共计18 个样本进行16S rDNA测序、数据质控后,共得到有效序列783372条,初乳组和常乳组分别得到优化序列条数为392724 条和390648 条。如图1 所示,两组样本的优化序列长度均主要分布在401~440 bp,集中在421~440 bp,与设计引物扩增长度300 bp左右接近,可满足序列分析要求。

图1 样本有效序列长度分布Fig.1 Distribution of effective column length of sample order
在97%相似性水平下对所有样本序列进行聚类分析[12],结果见图2,初乳组和常乳组共注释出1176 个OTU,两组共有的OTU为669,各自独有的OTU分别为79 和428。初乳组共产生748 个OTU,常乳组共产生1097 个OTU,由此可知,常乳组OTU数远高于初乳组,两组水牛乳样本间存在大量共有的细菌菌群,且常乳组中细菌多样性高于初乳组。
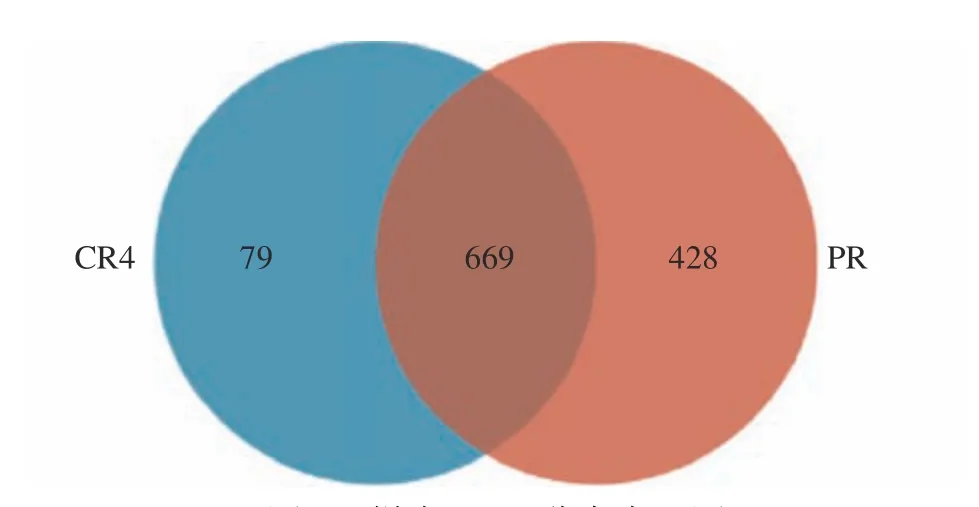
图2 样本OTU分布韦恩图Fig.2 Venn diagram of sample OTUs distribution
稀释性曲线主要反映测序深度,根据曲线是否达到平缓来判断本次测序数量是否足够,也间接反映样本中菌群的丰富度和多样性。Sobs和Shannon指数分别评估样本群落的丰富度和多样性[13]。样本基于OTU的Sobs和Shannon稀释曲线结果如图3 所示。各样本的群落丰富度Sobs指数稀释曲线大部分还在上升期,但群落多样性Shannon指数稀释曲线在3000 条时就已经平缓,说明增加测序数量可能会发现少量新的OTU,但样本群落多样性不会增加[14],说明本次测序数量足以满足后续数据分析要求。
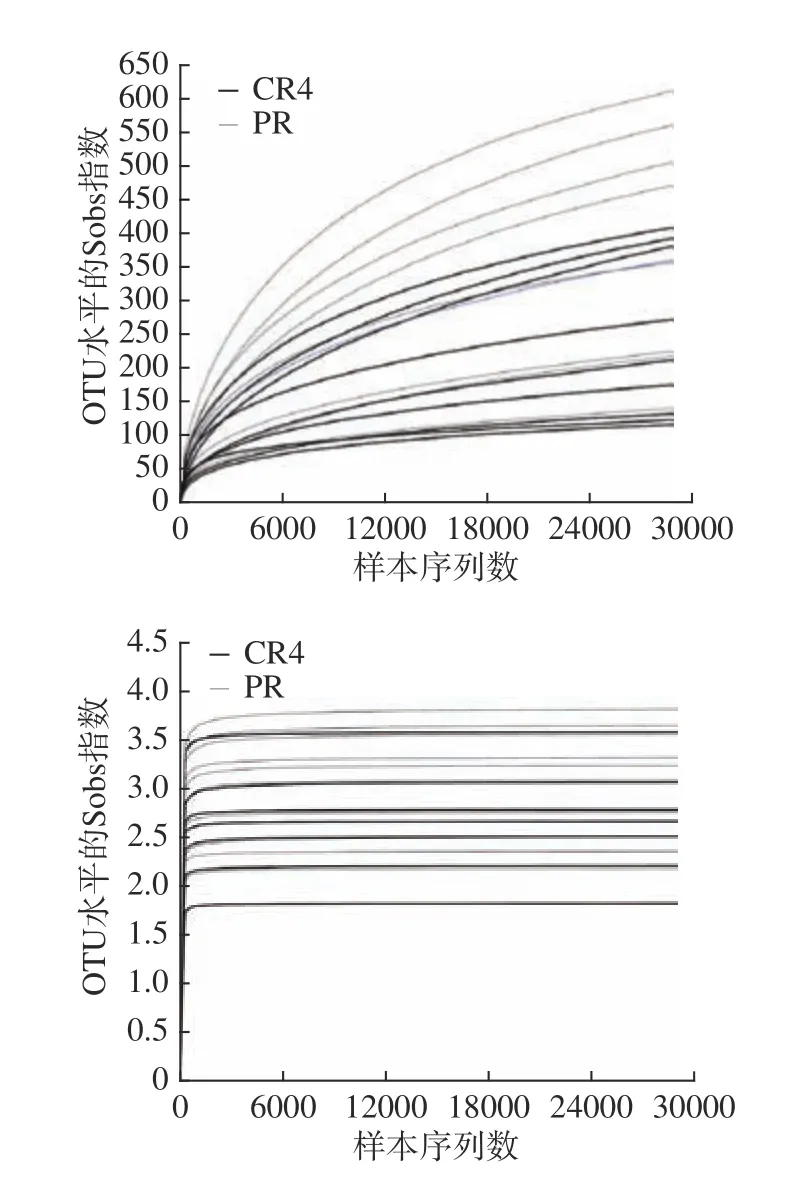
图3 Sobs(a)、Shannon(b)指数稀释性曲线Fig.3 Rarefaction curves for Sobs (a) and Shannon (b) index
2.2 细菌组成与丰度差异分析
基于OTU注释结果,分析样本在不同分类水平上的群落组成。本次测序,Coverage(覆盖率)为0.999,可代表99.9%的样本信息。两组样本共注释到1176 个OUT,26 个细菌门,97 个目,197 个科,495 个属和767 个种。初乳组样本共得到19 个门,52 个目,113 个科,292 个属和437 个种。而常乳两组样本共得到7 个细菌门,45 个目,84 个科,203 个细菌属和330 个菌种,相对而言,初乳组的细菌多样性高于常乳组。为可视化呈现各样本的物种丰度信息,本研究采用柱形图来展示各样本在门水平和属水平的菌落分布及差异性[15],结果如图4、图5 所示。由图4 可知,在门分类水平上,以相对丰度≥0.01%为阈值,将<0.01%的菌门归为others,初乳和常乳组共得到5 个细菌门,从高到低依次为变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、放线菌门(Actinobacteria )、Patescibacteri。其中变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)、放线菌门(Actinobacteria )在常乳组中占比较多,而拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)则在初乳组中较占优势,同一菌门在不同样本中的相对丰度差异较大。变形菌门、拟杆菌门、厚壁菌门为两组样本中的绝对优势菌门,相对丰度分别为35.4%、27.6%、24.8%,合计达88%。

图4 门水平菌落丰度图Fig.4 Abundance map of horizontal colony of phylum
在属水平上,丰度≥0.1%的细菌属有15 个(图5),其中含量较高的有9 个属:金黄杆菌属(Chryseobacterium)、不动杆菌属(Acinetobacter)、假单胞菌属(Pseusomonas)、巨型球菌属(Macrococcus)、栖水菌属(Enhydrobacter)、乳球菌属(Lactococcus)、肠球菌属(Enterococcus)、苍白杆菌属(Ochrobactrum)、链球菌属(Streptococcus),各属的相对丰度依次为24.7%、9.1%、6.6%、5.2%、5.2%、5.0%、4.7%、3.4%、2.9%,丰度和为61.6%,另外6 个属的丰度和为25.2%,其它<0.1%的菌属归为others,丰度为13.2%。
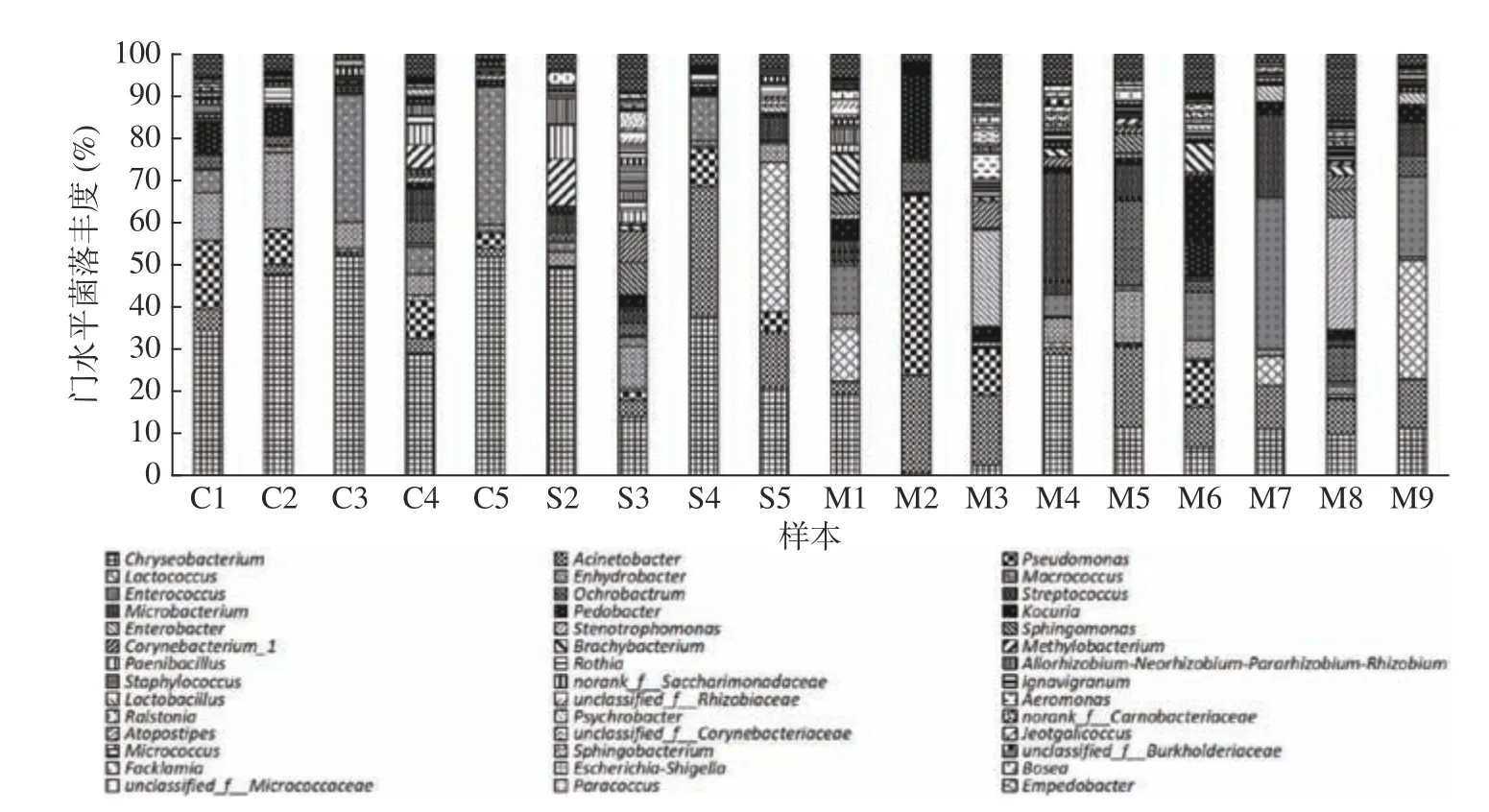
图5 属水平菌落丰度图Fig.5 Abundance map of horizontal colony of genus
由图5 可知,不同样本中主要菌属占比差异较大,相对而言,常乳组样本中菌群多样性要高于初乳组。在种水平上,将初乳和常乳两组样本中各自的同名菌种丰度求和,其占比如表1 所示,两组样本间优势细菌丰度差异较大。初乳组中的炭疽金杆菌(Chryseobacterium anthropi)、溶酪大球菌(Macrococcus caseolyticus)、奥斯陆莫拉菌(Moraxella osloensis)占比较大,而这几种菌在常乳组中的占比相对较少,尤其是炭疽金杆菌和溶酪大球菌。常乳组中占优势的菌为意大利肠球菌(Enterococcus italicus)、乌尔辛不动杆菌(Acinetobacter ursingii)、炭疽金杆菌(Chryseobacterium anthropi)。而别雷斯不动杆菌、假单胞菌、乔斯特金杆菌这三株菌在初乳组样本中的相对丰度较高,但在常乳组样本中却未检出或相对丰度小于<0.1%。

表1 丰度较高的细菌及其在门水平上的归类Table 1 Bacteria with high abundance and their classification at phylum level
在属分类水平,取样本平均总丰度前30 的菌属,按其物种和样本层级聚类方式绘制群落丰度聚类热图(图6),使高丰度和低丰度菌落分区聚集[16],通过颜色梯度变化来反映不同分组样本在属水平群落组成的相似性和差异性。由图6 可知,初乳组和常乳组整体并没有分区聚集,但两组样本中优势菌群的丰度明显不同,金黄杆菌(Chryseobacterium)、巨型球菌属(Macrococcus)、栖水菌属(Enhydrobacter)在初乳组样本中的含量显著高于常乳组,而假单胞菌属(Pseudomonas)、苍白杆菌属(Ochrobactrum)、肠球菌属(Enterococcus)在常乳组中的丰度均高于初乳组。金黄杆菌(Chryseobacterium)和不动杆菌(Acinetobacter)在两组样本中均占比较高,葡萄球菌属(Staphylococcus)在两组样本中均有低丰度出现。

图6 样本基于属水平的菌落聚类热图Fig.6 Community clustering heatmaps of samples based on genus level
2.3 细菌多样性比较分析
采用样本层级聚类树来分析初乳和常乳组间群落组成的相似性。18 个样本基于OTU的样本层级聚类树如图7 所示,不同颜色代表不同分组,树枝长度和宽度分别表示样本间的距离和远近,相似度越高,距离越小[17]。通过样本聚类树可以清楚地看出样本间分支距离远近,直观反映出各样本间菌群的相似度和差异性[18]。由图7 可知,除去个别异常样本,初乳组样本(红色)主要聚成一类,相似度较高,其中C1 和C2 是最相似的两个样本。常乳组(蓝色)则分成两类,其中M2、M3、M6 聚成一类,而M1、M5、M7、M8、M9 聚成另一类,其中M7、M9 相似度最高,这两聚类样本间相似度差异较大。整体而言,初乳组与常乳组组间样本差异不明显,初乳组组内样本间菌落组成相似度较高,而常乳组组内样本间菌落组成差异较大。

图7 样本层级聚类分析Fig.7 Sample level cluster analysis
对初乳(CR4)与常乳(PR)样本中细菌属(相对丰度阈值>0.1%)进行主成分(PCA)分析,以便找出两组样本间菌群组成的相似性和差异性。样本通过降维后,在主成分上会显示相对坐标点,样本点越接近,表明两样本物种组成越相似[19],同理,各点之间距离越大,表明它们之间的菌群差异越大[20],由图8 可知,初乳(CR4)与常乳(PR)两组样本代表的点都按各自分组相对聚集,但两组样本点之间的距离较近,在空间上没有完全分开,这说明初乳组与常乳组组间样本个体之间菌群组成有一定的差异,导致少部分样本菌群结构相似,大部分样本菌群存在差异性。主成分1(PC1)和主成分2(PC2)对样本差异性的贡献分别达到了41.21%和17.66%,共计58.87%,可以代表大部分变量信息。由图8 可知,初乳组与常乳组在第一主成分PC1 所在的X轴方向,两组样本的微生物组成有明显的分离趋势,这说明PC1 因素有较高的可能性影响了样本的组成,表明结果数据组内重复性好,组内样本差异性大,且PC1 贡献值为41.21%,可以作为有效的主成分,来解释两组样本的组间差异。
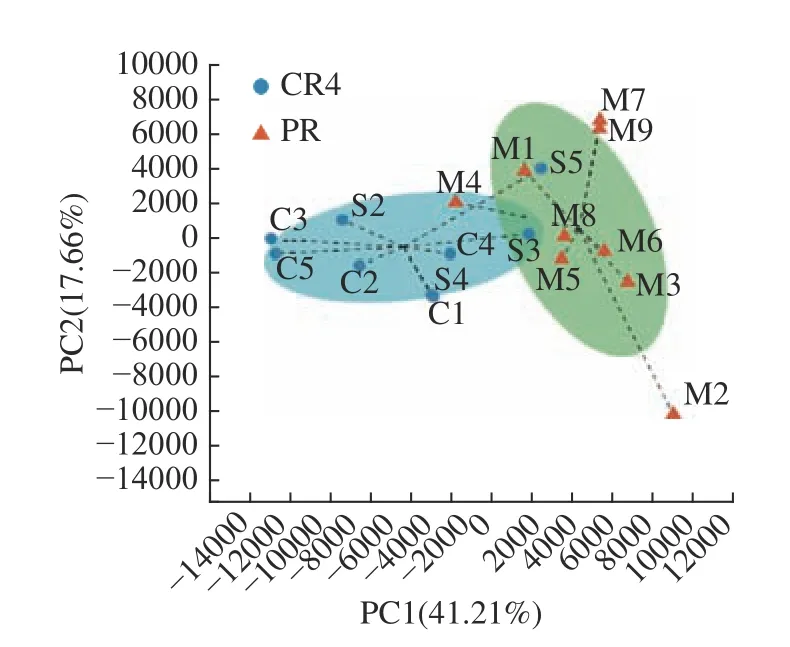
图8 样本基于属水平的主成分分析Fig.8 The sample principal component analysis on the genus level
PLS-DA(偏最小二乘法判别分析Partial Least Squares Discriminant Analysis)也是一种常见的检验样本组间差异的方法,此方法是根据分组方案,弱化组内样本差异,突出组间样本差异[21],更便于发现组间样本的差异性。由PLS-DA(图9)显示,除去M8、S3 两个离群严重的异常样本,其余样本点均按初乳与常乳所代表的分组相对聚集,两组样本点在COMP1(X轴)和COMP2(Y轴)方位能够很好地区分开来,说明初乳与常乳两组样本在组间的微生物组成存在明显差异。这种一目了然的分离效果在之前的PCA分析图中是没有得到体现的。初乳组(CR4)所代表的样本点都比较集中,说明初乳组组内样本之间菌群组成较相似,而常乳组(PR)所代表的样本点都比较分散,这说明常乳组组内样本间菌群组成有一定的差异性。结合PCA和PLS-DA可知,初乳与常乳组组间差异显著,组内差异不明显。

图9 样本基于属水平的PLS-DA分析Fig.9 Sample PLS-DA analysis based on the genus level
3 讨论
相较传统的涂片、划线等细菌培养和API试剂条等其它检测方法,16S rDNA高通量测序更全面和准确地反映了样本中菌群的多样性和丰度信息,解决了以往不可培养及低丰度菌在传统方法中无法分离鉴别的局限[22]。依据测序结果,本次试验Coverage(覆盖率)为0.999,显示样本细菌99.9%的信息得到了反映,且样本的Shannon稀释性曲线都趋于平缓,说明本次实验的测序深度合理,测序量足够,可代表样本中绝大数的菌群多样性信息。
本次测序,在门水平上,初乳和常乳组主要分属于变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)和厚壁菌门(Firmicutes),在属水平上,初乳和常乳组主要菌群种类不尽相同,初乳组中的主要菌群有金黄杆菌属(Chryseobacterium)、不动杆菌属(Acinetobacter)和假单胞菌属(Pseudomonas),而常乳组中的优势菌属主要为金黄杆菌属(Chryseobacterium)、乳球菌属(Lactococcus )和肠球菌属(Enterococcus)。黄卫强[23]在研究母乳微生物群落构成中,鉴定出的优势菌门与本试验结果相同,而在属水平上的差异较大。这可能是因为人乳和水牛乳因品种和乳源不同所致。Mazmanian等[24]的研究表明,Bacteroides(拟杆菌)在人类初乳中含量非常丰富,它可能在新生儿肠道早期定群中发挥主要作用。同样,本次试验水牛初乳样本中,拟杆菌的占比也较大。
研究[25−26]表明,金黄杆菌、不动杆菌、假单胞菌属均具有一定的抗感染能力,且假单胞菌属和肠球菌属与糖类和酶类代谢存在重要联系。综合本实验结果,水牛初乳和常乳组所拥有的这些核心菌属及其所具备的抗感染、修复能力以及糖和酶代谢功能,不论是对奶水牛产后乳房炎的修复还是犊牛早期肠道菌群定殖、消化等,都起到重要作用。
不论是在初乳还是常乳组中,炭疽金杆菌Chryseobacterium anthropi均被检测到占有较高比例(见表1),而炭疽杆菌属于需氧芽孢杆菌属[27],能引起人畜共患的炭疽病,牛、羊、骆驼等草食动物是其主工传染源[28]。炭疽杆菌对社会公共卫生和经济发展的危害,迄今仍占相当大的比重[29]。通过此次实验,说明水牛所种畜牧场大部分奶水牛可能或已经感染了炭疽病,应马上采取针对性的防治措施应对。
同样是水牛乳样本,在本实验检出的部分低丰度菌群,在某些研究中却是优势菌群[30],有研究[31−32]显示,水牛乳中Staphylococcus(葡萄球菌)、Streptococci(链球菌)在阳性组中所占比例较大,其组合感染是引起奶水牛乳腺病变的主要原因。本试验在两组样本中均检测到低丰度的Staphylococcus(葡萄球菌)和Streptococci(链球菌),尽管这些奶水牛在样本采集时并没表现出乳腺炎的病症[33]。这将为本所种畜厂奶水牛乳腺炎针对性的预防治疗提供依据。
4 结论
采用16S rDNA高通量测序技术,对广西水牛研究所种畜厂内的三品杂奶水牛初乳与常乳组样本中的细菌组成及多样性进行了比较分析,结果显示,两组样本共注释到1176 个OUT,26 个细菌门,97 个目,197 个科,495 个属和767 个种。其中初乳组样本共得到292 个细菌属和437 个细菌种。初乳组较常乳组细菌多样性丰富。通过PCA和PLSDA分析显示,初乳与常乳两组样本,组内差异较小,组间差异明显,各样本优势菌群和丰度差异较大。通过本次测序,得知了水牛研究所种畜场三品杂水牛乳中的菌群分布状况,对后续改善该种畜场奶源卫生条件、防止原料乳污染、乳腺炎的前期预警等均有指导意义。
