超高效液相色谱-串联质谱法测定茶叶中的百草枯
2021-07-21刘相真石琳王静叶美君
刘相真,石琳,王静,叶美君
(中华全国供销合作总社杭州茶叶研究院,浙江杭州310016)
百草枯(Paraqua)是一种速效触杀型灭生性季铵盐类除草剂,具有广谱速效和触杀作用,对绿色植物有很强的破坏作用,并且具有在土壤催化下快速失活的特点,失活后无毒素残留,对环境影响极小[1],在农业中被广泛推广使用。目前,百草枯的使用遍及全世界100多个国家和地区[2]。但是如不按照指导使用,百草枯对人畜有很强的毒性[3],且无特效解毒药,是导致人类急性中毒死亡率很高的除草剂[4]。由于百草枯具有高毒性和广泛使用性的特点,因此建立一种快速、高效、高灵敏度的检测方法至关重要。
目前,百草枯的检测方法主要有气相色谱(GC)[5]或气相色谱-质谱法(GC-MS)[6-7]、高效液相色谱法(HPLC)[8-11]或高效液相色谱-串联质谱联用法(HPLC-MS/MS)[12-14]、分光光度法(UV)[15-16]等方法[17-18]。气相色谱或气相色谱-质谱法具有分离效果好、灵敏度高、选择性强等优点,是分析检测中较常用的方法,但在百草枯检测中存在极性强、难以汽化需衍生以及前处理操作复杂等问题;高效液相色谱法是百草枯检测的常用方法,一般用反相色谱柱或亲水性色谱柱分离,可保留碱性化合物、无需使用离子对试剂,但存在灵敏度不高,对本底复杂样品定性不准等问题;超高效液相色谱串联质谱技术作为近年来得到广泛应用的技术,具有高选择性、高灵敏度和痕量检测等特点,成为检测分析的重要技术。
百草枯为强极性化合物,极易溶于水,采用水浸法提取效率高;在酸性、中性溶液中稳定,在碱性溶液中易分解,提取液中加入一定量的酸,可防止百草枯的降解。现有文献报道主要针对生物样本(血液或尿)、环境样品(水或土壤)及蔬菜水果,而茶叶基体复杂,含有大量酚类、糖类及生物碱,对质谱的雾化电离有着较大的抑制作用。试验基于超高效液相色谱串联质谱法,在SN/T 0293—2014《出口植源性食品中百草枯和敌百快残留量的测定液相色谱-质谱/质谱法》方法的基础上,针对茶叶样品,进一步优化前处理、色谱条件等,建立了一种准确、快速、高灵敏度的测定方法,且测定结果满足各项质控要求。
1 材料与方法
1.1 试验材料
市售茶叶样品红茶、绿茶、普洱茶和乌龙茶,茶叶磨碎样品按照GB/T 8303—2013制备。
1.2 仪器与试剂
UPLC/TSQ Quantum Access MAX超高效液相色谱-三重四极杆质谱联用仪 (美国Thermo Fisher-scientific公司);分析天平(感量0.0001 g,瑞士Mettle Tolede公司);FJ-12固相萃取装置(上海京孚仪器有限公司);SC-3610低速离心机(安徽中科中佳科学仪器有限公司);漩涡混合器(海门其林贝尔仪器制造有限公司)。
甲醇(色谱纯,美国Tedia公司);甲酸铵和乙酸铵(色谱纯,美国Fluka公司);甲酸和37%盐酸 (色谱纯,德国Merck公司);百草枯标准品(100μg/mL,AccuStandard,Inc);200 mg/3 mL C18SPE Cartrideg(岛津技迩商贸有限公司);60 mg/3 mL WondaSepWCX固相萃取柱(岛津(上海)实验器材有限公司);60 mg/3 mL Poly-sery MCX固相萃取柱(ANPEL Laboratory Technologies(Shanghai)Inc);60 mg/3 mL Poly-sery HLB固 相 萃 取 柱(ANPEL Laboratory Technologies(Shanghai)Inc)。
1.3 标准溶液的配制
准确移取1 mL百草枯标准品,用甲醇溶解并配制成质量浓度为10μg/mL的标准储备液,于4℃冰箱中避光保存。使用时,用甲醇-水-甲酸溶液(29∶70∶1,V/V/V,含0.5 mol/L甲酸铵)逐级稀释成5、10、25、50、100、200μg/L标准系列溶液。
1.4 样品前处理
1.4.1 提取
称取磨碎茶样5.0 g(精确至0.001 g)置于50 mL具塞聚乙烯离心管中,加入20 mL甲醇-0.1 mol/L盐酸溶液 (3∶7,V/V), 旋涡提取1 min后,再超声提取15 min,以4200 r/min离心5 min,取上清液至50 mL容量瓶中,残留物继续加入20 mL甲醇-0.1 mol/L盐酸溶液(3∶7,V/V)重复提取一次,合并两次提取液于同一容量瓶中,并用水定容至刻度,摇匀,待净化。
1.4.2 净化
移取10 mL提取液过预活化后的MCX强阳离子固相萃取柱,弃去流出液;分别用2 mL水、乙腈-甲酸溶液(99∶1,V/V)和甲醇淋洗,然后置于SPE固相萃取装置-真空泵系统中,将残留的试剂抽干。 最后用3 mL甲醇-水-甲酸溶液(29∶70∶1,V/V/V,含0.5 mol/L甲酸铵)洗脱,收集洗脱液于5 mL塑料离心管中,旋涡混匀后,过0.22μm有机滤膜,待测。
1.5 仪器条件
1.5.1 色谱条件
色谱柱为Thermo SyncronisTMHILIC(100 mm×2.1 mm,1.7μm,赛默飞世尔科技);流动相A为0.2%甲酸水溶液(含50 mmol/L甲酸铵),流动相B为甲醇,梯度洗脱条件见表1;柱温40℃;进样量2.0μL。
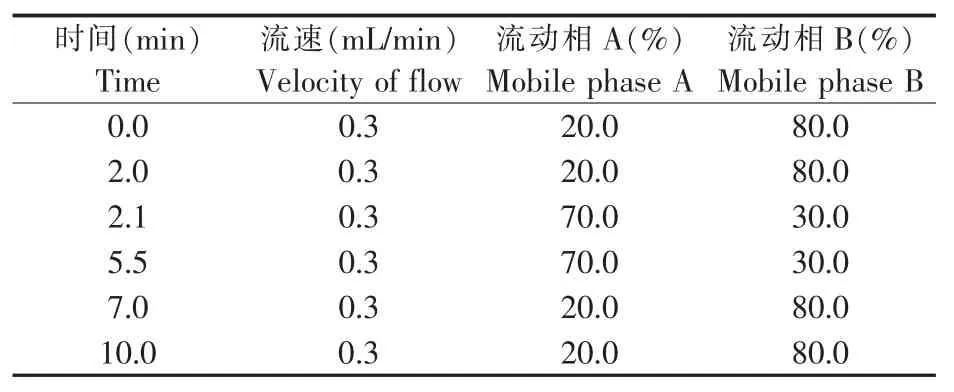
表1 梯度洗脱程序Table 1 Program of the gradient elution
1.5.2质谱条件
电离模式:电喷雾离子化(ESI);电离源极性:正模式;雾化气:N2;离子喷雾电压:4000 V;雾化室温度120℃;离子传输管温度380℃;碰撞气:Ar,0.12 Pa;扫描模式为SRM多反应监测扫描,扫描时间段为3.5~5.5 min。质谱参数见表2。

表2 百草枯的质谱参数Table 2 MS/MS parameters for paraqua
2 结果与分析
2.1 前处理条件优化
2.1.1 提取溶剂的选择
百草枯极易溶于水,用纯水、0.1 mol/L盐酸溶液、甲醇-0.1 mol/L盐酸以及甲醇对茶叶中的百草枯进行提取,发现单独用纯水、0.1 mol/L盐酸溶液、甲醇对茶叶中百草枯的提取率不高,通过在盐酸溶液加入一定比例的甲醇可以显著提高提取率。分别对纯水、0.1 mol/L盐酸溶液、甲醇以及不同比例的甲醇-0.1 mol/L盐酸溶液 (1∶9、2∶8、3∶7、4∶6、5∶5,V/V)的提取效率进行了比较,结果如图1所示。甲醇-0.1 mol/L盐酸溶液(3∶7)提取回收率最高,为96%,故选为试验的提取试剂。
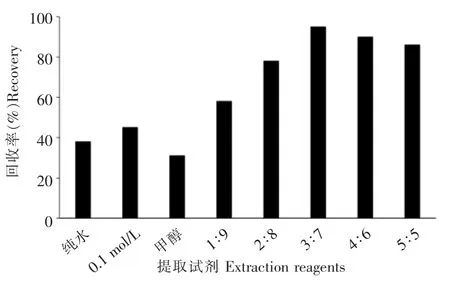
图1 不同提取试剂对茶叶中百草枯的回收率比较Fig.1 Comparison of recoveries with different extraction reagents for paraquat in tea
2.1.2 净化方式的选择
百草枯在酸性溶液中主要以离子形式存在,可采用离子交换固相柱净化。试验对C18(疏水性反相硅胶基质吸附柱)、HLB(亲水性反相吸附柱)、MCX(强阳离子交换柱)和WCX(弱阳离子交换柱)的净化效果进行了比较。发现用C18和HLB净化,茶叶中的茶多酚、咖啡碱及色素等均无法除去,杂质较多,影响质谱检测的灵敏度,检出限为0.1μg/mL,无法满足质控的需要;用WCX净化,在过柱前需用1 mol/L NaOH溶液调节提取液pH值至7.0±0.1,茶叶提取液会产生絮状沉淀影响过柱效果,百草枯回收率较低,特别在低浓度时,0.01、0.05μg/mL的回收率分别仅有32%和60%,造成该结果的原因可能是加入NaOH溶液打破了原有提取液的电解质平衡,在离子效应、电解质作用和共沉效应等共同作用下,茶多酚、氨基酸、糖类等聚合成大分子物质形成络合物而沉淀,影响百草枯的回收率。
由于百草枯的pKa大于10,MCX柱在酸性条件下对百草枯有很强的相互作用力,可以直接在酸性条件下直接过柱,无需调节pH值,并且可以用水、甲醇、乙腈-甲酸溶液(98∶2,V/V)或氨水-甲醇(5∶95,V/V)、氨水-乙腈(5∶95,V/V)淋洗柱子去除茶多酚、咖啡碱、糖类及色素等杂质,且不会造成百草枯的损失,达到较好的净化效果;采用离子强度更大的甲醇-水-甲酸溶液洗脱,洗脱强度随着甲酸铵缓冲液的浓度增大而增大,洗脱体积随之减少,但考虑高浓度的盐易造成色谱柱堵塞和对质谱离子化效果的减弱,试验采用甲醇-水-甲酸溶液(29∶70∶1,V/V/V,含0.5 mol/L甲酸铵)实现了较完全洗脱,其洗脱效果如图2所示。
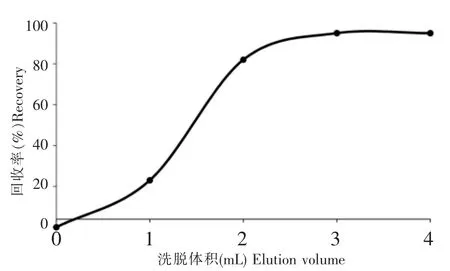
图2 百草枯标准溶液在MCX小柱上的洗脱曲线Fig.2 Elution curve of paraquat on MCX column
2.2 检测条件优化
2.2.1 质谱条件的优化
根据百草枯的结构特征,采用ESI+模式。用10μg/mL百草枯的单体标准溶液进行质谱条件优化,选择准分子离子[M+H]+作为母离子,进行MRM模式优化各质谱参数(包括碰撞能量、透镜补偿电压等),以获得稳定性好、信号强度高的碎片离子。100 ng/mL的百草枯提取色谱图、空白茶叶样品提取色谱图和茶叶样品中添加0.3 mg/kg的百草枯提取色谱图如图3所示。

图3 百草枯的质谱图Fig.3 MRM chromatograms of paraquat
2.2.2 色谱柱的选择
由于百草枯极性较强,在反相色谱柱上的保留时间极短,难以与干扰物有效分离。实验选用Thermo SyncronisTMHILIC色谱柱进行分析,能够有效实现百草枯的保留以及与干扰物的分离。
2.2.3 流动相的选择
试验考察了有机相乙腈与水相中不同pH值的甲酸水溶液和不同浓度的甲酸铵对百草枯的分离效果的影响,结果表明,水相中不同浓度的甲酸铵含有的甲酸浓度低于0.2%时,峰型裂峰或拖尾,并且检测灵敏度低;在水相中添加0.2%甲酸时,百草枯的灵敏度高,分离效果满意。可能因为在正离子模式下,酸性能够促进化合物的离子化,提高化合物的离子化效率和灵敏度。
试验在0.2%的甲酸水溶液的基础上,进一步添加不同浓度的甲酸铵。结果发现,在一定范围内,随着甲酸铵浓度的升高,峰宽变窄,峰高升高,信噪比增大,灵敏度提高,峰型对称性变得更好。当甲酸铵浓度大于50 mmol/L时,灵敏度无明显提升,但高浓度的甲酸铵会造成泵的损坏、色谱柱和管路系统的堵塞。因此,实验选择甲醇0.2%甲酸-甲醇溶液(含50 mmol/L甲酸铵)作为流动相。
2.3 方法验证
2.3.1 线性关系和方法检出限
按“1.3标准溶液配制”和“1.5仪器条件”进行测定,根据3倍信噪比(S/N)确定百草枯的检出限为3μg/kg,10倍信噪比(S/N)确定百草枯的定量限为10μg/kg。百草枯在标准系列范围内峰面积与样液浓度呈线性相关,百草枯的计算回归方程为y=0.0197x-0.0179,R2=0.9994,线性关系良好。
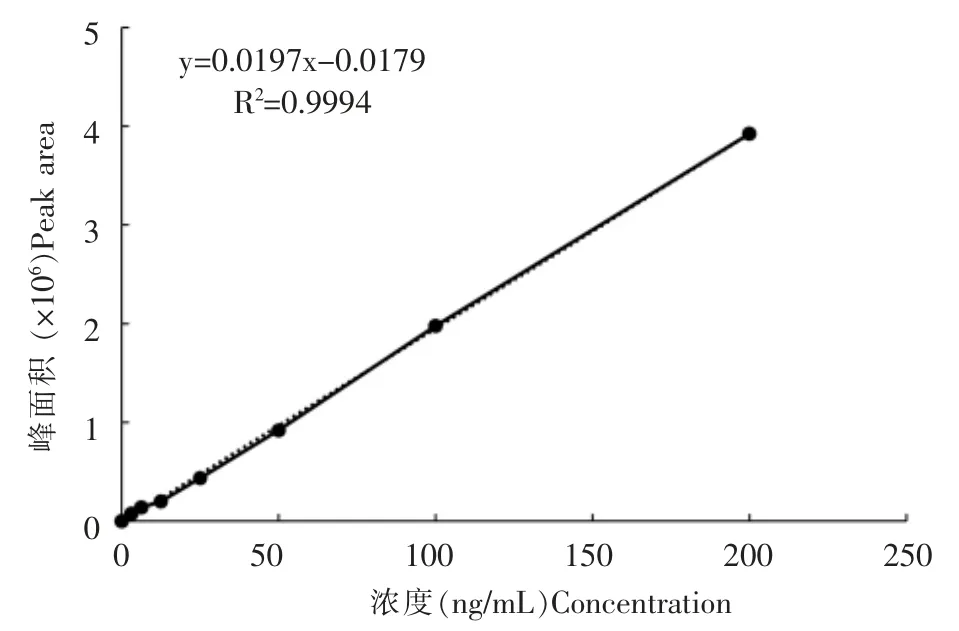
图4 百草枯的标准曲线(0.005~0.200μg/mL)Fig.4 Standard curve for paraquat(0.005~0.200μg/mL)
2.3.2 准确度和精密度
选取市售绿茶、红茶、普洱茶、乌龙茶样品,分别作空白及添加相当于定量限、2倍定量限及0.2 mg/kg的标准品,按上述方法进行分析检测,采用标准工作曲线外标法定量,每个加标水平平行测试6次,加标回收率与精密度如表4所示。结果显示,方法的回收率为81.4%~93.5%,相对标准偏差为2.1%~5.6%,其准确度与精密度均能满足茶叶中百草枯检测的要求。
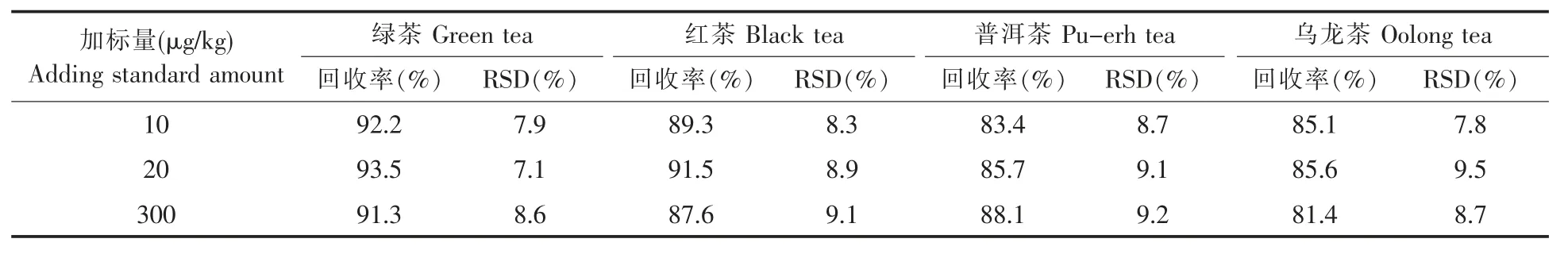
表4 茶叶中百草枯的加标回收率和精密度Table 4 Recoveries and relative standard deviations(RSDs)of paraquat in tea
2.3.3 基质效应
为考察茶叶的基质效应,以空白绿茶、红茶、普洱茶、乌龙茶为基体,按照1.4所述方法进行前处理,用收集的基质配置标准系列溶液制成标准曲线,与以无基质配制的标准曲线作对比。结果显示,以空白绿茶、红茶、普洱茶、乌龙茶为基质的标准曲线的斜率与无基质的标准曲线的斜率的比值分 别 为1.03、1.12、1.05、1.08, 其 斜 率 比 值 均 在0.8~1.2之间,认为基质效应较低[19],故文章采用甲醇-水-甲酸溶液(29∶70∶1,V/V/V,含0.5 mol/L甲酸铵)配制的标准曲线进行定量。
3 结论
茶叶中的化学成分复杂,含有色素类、生物碱、多酚类等物质会干扰样品的检测,对检测结果造成影响。文章根据MCX柱填料与百草枯具有很强的相互作用力,采用在酸性条件下,提取后直接过柱,用水、乙腈-甲酸溶液和甲醇淋洗柱子去除大部分色素、多酚等物质后,用甲醇-水-甲酸溶液(含0.5 mol/L甲酸铵)洗脱,操作简便、净化效果好,且准确度与精确度均能满足植物源性食品中农药残留检测的需要,可作为农药残留分析的日常检测方法。
