全外显子组测序在自身免疫性疾病中的研究进展
2021-07-15马晓敏王欣姚新生谭贵琴周芳宇魏文文于红松
马晓敏王 欣姚新生谭贵琴周芳宇魏文文于红松
(遵义医科大学基础医学院,贵州遵义 563000)
自身免疫性疾病(autoimmune diseases,AIDs)是一类因免疫功能发生紊乱或机体自身发生免疫应答从而使得自身器官系统受损的疾病[1]。 正常机体的免疫系统具有区别“自己”和“非己”的能力,对非己抗原能够发生免疫应答,对自身抗原则是处于无应答或微弱应答的免疫耐受状态;如果正常的免疫耐受被打破,处于非正常免疫激活状态的T 细胞就会持续迁延,对自身抗原产生异常的免疫应答,导致AIDs 的发生;AIDs 的主要特征是患者血清中可检测到高浓度的自身抗体或是能够与自身组织起反应的淋巴细胞。 研究发现,环境因素和遗传因素可以增加普通人群患AIDs 的风险[2-4]。 大量遗传学研究还表明,不同的 AIDs 有着共同的遗传背景[5-7]。
全基因组关联研究(genome-wide association studies,GWAS)、 全 外 显 子 组 测 序 (whole exon sequencing,WES)与全基因组测序等技术的飞速发展,极大地加深了我们对AIDs 相关遗传因素的理解[8]。 GWAS 的核心是研究分子变异和目标表型性状之间的关联,极大地扩大了对最常见形式AIDs 遗传因素的理解,但只能部分解释复杂性状的遗传性,不能找到真正的致病基因或者突变位点,对基因与环境因素交互作用缺乏足够的关联研究,GWAS 对低频变异和罕见变异的检测能力也十分有限[8-11]。 全基因组测序是针对个体所有基因序列进行的测序分析,其测序范围广、数据多、分析困难、专业性强、费用昂贵,因此目前难以大规模应用于临床[12]。 而WES 则只是针对DNA 的外显子区域,更简便、经济和高效,同时目标区域覆盖度更高,便于变异检测;虽然WES 捕获探针仅覆盖人类基因组的1%~1.5%的区域,但是该区域包含了大约85%的已知致病基因变异,可一次性精准检测人类基因组20000 多个基因中180000 个外显子的致病变异位点;WES 还解决了GWAS 对罕见突变和结构变异性不敏感的缺点,通过其高效的方法能鉴定新的稀有编码变体[13-14]。 本文主要以系统性红斑狼疮、类风湿性关节炎、多发性硬化病、银屑病、干燥综合征、白塞病等常见AIDs 为例,对 WES 在 AIDs 中的应用及研究进展进行综述。
1 WES 与系统性红斑狼疮
系统性红斑狼疮(systemic lupus erythematosus,SLE)是一种累及多系统的慢性AID,病程迁延反复[15]。 邝少松等[16]通过小鼠动物模型证实其主要的病理特征是T 淋巴细胞的失衡,导致了机体免疫紊乱产生大量自身抗体引发炎症和组织损害,常累及全身多系统或多器官病变,包括心脏、各个关节、皮肤、肺、血管、肝、肾,以及神经系统;在我国人群中的患病率为70/10 万,全国患病总人数近一百万,尚缺乏特异的有效治疗手段[17]。 GWAS 发现SLE存在遗传易感性,目前已发现SLE 发病与多个易感基因有关[18]。
SLE 的发病可能是机体免疫失衡所致,近年来,研究人员通过WES 技术已经新鉴定出多个遗传变异可能通过打破免疫耐受参与SLE 的发生。 李国民等[19]对一例SLE 患儿及其父母进行了WES 和生物信息学分析,后使用Sanger 法验证突变位点并在其家系其他成员中进行突变分析,筛选得到92 个变异位点,检测到1 个可能致病基因,即SLC7A7 经典剪切区IVS4+1 G>A 纯合变异,此基因突变可以引起巨噬细胞和淋巴细胞中氨基酸代谢异常,从而导致免疫耐受被破坏,出现自身抗体。 李国民等[18]对另一例SLE 患儿及其父母进行WES,发现了105 个变异位点,筛选到可能的致病基因NRASc.38G>A 杂合突变。 而Li 等[2]对7 例SLE 患者及其家属进行WES,在4 例患者外周静脉血中发现了杂合的NRASc.38A>G 突变,但其父母均未携带,故为新发突变;另 2 例患者检测到TNFAIP3c.559C>T 和PIK3CDc.3061G>A 杂合突变;NRAS基因编码蛋白是RAS/MAPK 信号通路中的信号转导分子之一,该信号通路可将细胞外生长因子信号传至胞内,对调节细胞周期和细胞的分化、生长、衰老及凋亡起重要作用,因此该基因杂合突变会使RAS/MAPK 信号通路调节异常[17];TNFAIP3 是一个系统的负调控因子,主要诱导免疫耐受,其杂合突变可能打破机体免疫平衡,导致AIDs 的发生[20];PIK3CD编码的磷脂酰肌醇3-激酶(PI3K)在免疫细胞的激活和功能中起关键作用,基因突变引起的PI3K 活性增强会导致AIDs 的发生[21]。 此外,有研究发现相关基因的突变也可以通过调节B 细胞相关信号通路导致AIDs 的发生,Al-mayouf 等[22]对一例罹患 SLE 的六岁女童进行WES,分析确定了致病性TALDO1c.793Cdel 纯合子变异和PTENc.518G>C 杂合变异;PTEN可通过B 细胞受体调节正常信号,并在建立外周免疫耐受中发挥作用,其杂合变异可能诱导AIDs 的发生。 Jiang 等[23]对 116 例 SLE 患者 行WES,对97 名没有慢性病病史的健康老年人行GWAS,研究鉴定出BLK基因6 个罕见突变或新的错义突变以及BANK1 基因的低频突变参与了SLE的发病。
以上研究表明,通过WES 可以检测到与SLE相关的基因变异位点,这些突变位点通过打破自身免疫耐受、影响信号通路等不同机制使得机体免疫失衡而导致SLE 的发生,这促进了我们对于SLE 发病遗传因素的认识,也可能为SLE 的诊断与治疗提供靶标,但尚有一些突变位点缺乏对其相应机制的阐释,还需进一步研究来证实。
2 WES 与类风湿性关节炎
类风湿性关节炎(rheumatoid arthritis,RA)是一种病因不明的慢性多发性AID[24],其基本病理改变为滑膜炎、血管翳的形成,并逐渐出现关节软骨和骨的破坏,最终可导致关节畸形和功能障碍,影响了高达1%的世界人口,由遗传和环境危险因素共同作用导致其发生,其中遗传率约为65%[25-26]。 最近的GWAS 和相关性研究已经确定了50 多个RA易感位点,GWAS 的模拟数据和直接测序数据表明,高频、低频和罕见变异体的混合导致了RA 患病风险的上升[27]。 近年来,大量研究人员已将WES 技术应用到对RA 遗传因素的研究中。
Okada 等[28]通过 WES,在人类染色体 2q23 区域的PLB1 上发现了一个非同义的c.2263G>C(p.G755R)突变,该突变在显性遗传模式的家系成员中与RA 显著共分离,随后在1088 例RA 患者和1088名健康对照者(欧洲血统)中进行PLB1 基因的测序分析,证实该突变的存在及其显著致病风险。 Wang等[29]对51 例不伴有间质性肺疾病(interstitial lung disease,ILD)的 RA 患者和 45 例合并 ILD 的 RA 患者(RA-ILD)进行 WES,研究发现在 RA-ILD 队列中,携带MUC5B变异的患者急性加重或死亡的风险更高,而RA 患者MUC5B突变率为17.6%,因此其变异体的携带者状态可作为RA-ILD 预后降低和病情加重的指标。
综上,WES 研究发现了新的遗传变异参与RA的发生,另外发现患者对相应变异体的携带与否和疾病的严重程度有关,这有助于临床对患者预后的判断,但由于WES 在RA 中的应用研究依然较少,更多变异位点尚需进一步的研究确定。
3 WES 与多发性硬化症
多发性硬化症(multiple sclerosis, MS)是一种炎症性、自身免疫性脱髓鞘疾病,以轴突损伤、神经元丢失和中枢神经系统萎缩为特征[30-31]。 越来越多的证据表明,MS 的发生由环境和遗传因素共同作用导致[32]。 近期的 GWAS 和 Meta 分析发现,人类白细胞抗原DR(HLA-DR)的多态性与欧洲和北美等地区的高加索人群MS 的易感性显著关联,而与包括中国人群在内的亚洲人群MS 的发生却无显著关联[30],遗传异质性表明MS 尚有其他关键的遗传变异未被鉴定,而WES 的应用丰富了我们对MS相关遗传因素的认识[33]。
Wang 等[30]对来自中国南方的8 例MS 患者和26 名健康对照者外周血单个核细胞中的基因组DNA 进行WES,共发现了41227 个可能的变异,有17 个变异体具有显著的等位基因频率,经Sanger 测序证实,3 例 MS 患者具有异质性变异,即位于TRIOBP第 7 外显子的罕见变异(Chr22 ∶37723520G>T,rs201693690),导致了氨基酸替换,MS 患者的此等位基因频率明显高于健康对照组,因此TRIOBP可能是中国南方人群 MS 的新的危险基因,但TRIOBP如何影响免疫或炎症的机制尚不清楚。Kattimani 等[31]对1 例复发性缓解性多发性硬化症(relapse-remitting multiple sclerosis, RRMS)女性患者进行 WES 分析,发现miR-8485 携带 CA 碱基的移码纯合子缺失;而NRXN1 携带外显子8 的CT 到TC 碱基的非移码纯合替换,丝氨酸替换为亮氨酸;这二者突变改变了钙稳态和NRXN1/NLGN1 细胞粘附分子结合亲和力,miR-8485 可诱导NRXN1 基因沉默,而NRXN1 在调节钙通道活性和突触处Ca2+触发的神经递质释放中发挥重要作用,因此突变体miR-8485 对NRXN1 调节表达失调,会导致NRXN1的过度表达,触发Ca2+内流增加,Ca2+持续升高可能引起氧化应激和细胞死亡,导致神经变性。
上述这些研究结果提示多个遗传变异参与了MS 的发生,且在不同人群中有所差异,另外还发现这些变异可以通过相应机制影响到患者的具体症状,对于研究相应治疗方式提供了新的思路,但是仍有变异位点的关键作用机制尚且不明,有待进一步的研究。
4 WES 与银屑病
银屑病是一种免疫介导的具有多基因遗传性和复发性的慢性炎症性皮肤病,以红细胞斑块、角质形成、细胞增生和淋巴细胞浸润为特征,病程较长,易复发,有的病例甚至终生不愈,其发病率约为世界人口的2%,且该病发病以青壮年为主,对人类的身体健康和精神状况均造成较大影响,常由感染、外伤、药物等环境因素诱发;GWAS 已鉴定了多个与银屑病相关的易感位点,但仍未能清楚解释其发病机理,而WES 技术的应用加深了我们对银屑病发病机制的理解[34-37]。
Signa 等[38]研究了一个以儿童期起病的红皮病型银屑病为特征的有3 对双胞胎的大家庭,对该家族的5 名成员进行了WES 分析;在CARD14 第4 外显子中发现新的杂合突变(C.446T>G),此突变可导致以弥漫性红皮病型银屑病为特征的不寻常的临床表型和严重的儿童期银屑病的发生,该变异的存在通过Sanger 测序得到证实;同时该研究发现不同的SNP 可对生物制剂的疗效造成影响,这将有助于我们对银屑病的精准治疗。 此外,还有研究发现有些突变与患者治疗疗效相关,Kuang 等[34]采用WES对22 例寻常型银屑病患者(11 例甲氨蝶呤治疗有效和11 例甲氨蝶呤治疗无效)进行了检测,研究发现3 个错义突变(SMG6/rs216195 T>C、IMMT/rs1050301 G>A 和UPK1A/rs2285421 T>C)可能与甲氨蝶呤的疗效有关,但其具体作用机理还需进一步研究证实。 Carlsson 等[39]研究发现,UPK1A在银屑病等炎症性皮肤病中表达显著升高,但是否由UPK1A/rs2285421 T>C 错义突变所导致尚不清楚。
以上研究表明,WES 研究新发现了一些参与银屑病发生的遗传变异,这些变异不仅仅导致疾病的发生,还有可能与治疗效果相关,但尚未阐明具体机制,亟待进一步更深入的研究。
5 WES 与干燥综合征
干燥综合征(Sjögren’s syndrome, SS)是一种以口眼干燥为典型表现的AID,随病情进展常累及全身各个系统器官[40]。 SS 可分为单独发病的原发性干燥综合征(primary Sjögren’s syndrome, pSS)和伴有其他 AIDs 的继发性干燥综合征(secondary Sjögren’ s syndrome, sSS),其病因复杂,具体发病机制尚不明确,目前普遍认为其发病机制涉及遗传因素、感染因素和内分泌因素等[41]。 近期的WES 研究,进一步加深了我们对SS 发病机制的理解。
杜蒙[40]应用WES 分析了91 例pSS 患者和152名健康对照者,筛选到25 个罕见变异位点,进一步分析发现,CREBBP和OAS1 是pSS 的易感基因,但其具体作用机制尚不清楚。 王启迪等[41]对11 例pSS 患者和6 名健康对照者进行WES,筛选获得25个候选变异位点,后通过Sanger 测序验证,结果表明,IFIH1c.2115A>C、SIDT1c.1216G>A 可能与 SS 发病相关;IFIH1 编码胞浆RNA 解旋酶,可在病毒感染过程中提供识别dsRNA 的胞浆受体,通过线粒体抗病毒信号蛋白激活IFN-I 信号通路;SIDT1 是一种多次跨膜蛋白,其膜外区域可结合 dsRNA,并将dsRNA 转运到细胞内,其作用可能主要与pSS 患者机体的多系统和器官累及有关;因此,推测IFIH1 可能是导致pSS 发生的主效基因,而SIDT1 仅为其中的一个微效基因,然而变异位点的影响、基因在疾病中的具体作用还需功能实验的证实。 Shen 等[13]分析15 例SS 患者和100 名健康对照者的WES 数据,确定了PRAMEF13、TARBP1、UGT2B28、TRBV56和NAPB为 SS 新的易感基因,并发现UGT2B28/rs72552705 和MSH5/rs2075789 为 SS 风险基因位点;UGT2B28 在调节类固醇激素方面起着重要作用,具有结合某些胆汁酸和雌激素的固有能力,类固醇激素影响免疫细胞功能和炎症,但其与AIDs 的关系缺乏数据验证。
WES 的应用不仅鉴定了SS 相关的新的遗传变异位点,还发现这些遗传变异位点在疾病的发生发展中起到了不同程度的影响,这为进一步阐释SS 的发病机制提供了新的思路和研究靶点。
6 WES 与白塞病
白塞病(Behçet’s disease, BD)是一种累及多系统的由自身免疫介导的炎症性疾病,以非特异性血管炎为主要病理损害,其典型的临床特征包括口腔和生殖器溃疡、葡萄膜炎和皮肤病变等,其发病机制尚不明确,目前普遍认为BD 的病因包括遗传学、表观遗传学、免疫学及环境因素等[42-44]。 近年来WES 研究鉴定发现了多个遗传变异参与BD 的发生发展。
Dimopoulou 等[42]对一例 BD 患者及其父母进行了WES,研究发现BD 患者ERAP1 基因启动子区域出现罕见的纯合子突变(1-bp 缺失),且BD 患者中ERAP1 的表达水平显著低于正常对照;此研究也发现WES 可以补充基于DNA 微阵列的GWAS 研究,以调查稀有和非编码变体。 Ognenovski 等[43]对14 例来自德国的 BD 患者进行了 WES,并通过Sanger 法测序验证WES 结果,研究发现了与BD 发生相关联的77 个变异体,其中2 个为新发现的关联变异体(LIMK2/rs149034313 和NEIL/rs5745908);LIMK2/rs149034313 影响趋化因子和细胞因子信号通路介导的炎症、T 细胞激活以及血管生成,还参与调控细胞骨架重组,而细胞骨架重塑是BD 中白细胞黏附和浸润的中心;NEIL1/rs5745908 是一个剪切位点变异,可能引入有害的内含子保留,导致终止密码子提前出现以及产生非编码转录本,这种有害的效应可能会对氧化应激后的DNA 损伤修复产生不良后果;由于这些基因对BD 中白细胞浸润和氧化应激的增加是至关重要的,因此所发现的罕见变异关联可能会为未来的研究提供新的疾病靶点和关键途径。 Shigemura 等[44]调查了一个有BD 病史的日本家庭,对6 例4 代以上BD 患者进行分析,对患者1 和他母亲(患者2)单个核细胞提取的基因组DNA 进行WES,研究发现BD 患者均携带TNFAIP3杂合错义突变(chr6_138197226_G>A,C243Y),该突变可能通过抑制NF-κB 的活化而增加人类炎性细胞因子(IL-1β, IL-6 和 TNF-α) 的产生,表明TNFAIP3 突变(chr6_138197226_G>A,C243Y)可能是常染色体显性BD 的致病基因。
目前,尽管WES 在BD 中的应用较少,但现有WES 研究已经鉴定多个基因变异参与BD 的发生,这些变异通过影响炎性细胞因子的生成、调控细胞骨架系统或增加氧化应激等机制参与了BD 的发生发展,这加深了我们对BD 发生相关遗传因素的认识,有利于将来对其具体发病机制的阐释并应用于临床诊断和治疗。
通过以上所述可知,WES 可以检测到与AIDs相关的基因位点变异(详见表1),但某些位点的变异不仅仅局限于一种疾病,多种AIDs 在遗传风险上也共享某些疾病易感基因(详见表2)。 例如,IFIH1作为介导Ⅰ型干扰素产生的抗病毒解旋酶基因之一,可导致干扰素调节因子和NF-κB 等转录因子的激活,从而触发先天免疫应答,这可能会增加AIDs的风险,经研究证实其遗传变异与多种AIDs 易感性有关联:rs1990760 为 SLE、RA、MS 等的共有风险变异,其与血清IL-18、颗粒酶B 水平呈显著正相关,IL-18 可调节T 细胞分化,打破机体免疫平衡,导致严重的组织炎症和自身免疫;颗粒酶B 通过促进抗原表位的产生和刺激自身反应性T 细胞在获得性免疫应答和自身抗原的呈递中起着重要作用,还可以促进 IL-18 的激活,而 IL-18 在 SLE、RA、MS 中均有异常增高。 此外,rs3747517 与 SLE、MS 的发病相关,独立SNPs 的稀有等位基因rs10930046 变异的携带对银屑病、MS、SLE 有保护作用[45-50]。 而多项研究发现,TNFAIP3 的遗传变异也与 SLE、RA、MS、银屑病、SS、BD 等 AIDs 相关,rs2230926 是 SLE 和SS 的共有变异位点,作为NF-κB 途径的重要负反馈调节因子,rs2230926 异常导致NF-κB 通路的失控,可能参与AIDs 的发生发展[51-52]。 而BD 的易感因子ERAP1 的变异位点rs30187 与银屑病和MS 的遗传易感性有关联,其能够调控MHC I 类分子的表达,还有助于释放炎性细胞因子的膜结合受体,通过对抗原的处理和提呈及影响肽修剪参与机体免疫应答[53-54]。MSH5 也被发现在 SLE、MS 及 SS 中发挥作用,其变异使得调节失调可能导致相关疾病易感性, 但具体机制还需更深入研究来证实[13,55-56]。 由此可见AIDs 共享的疾病易感基因可能对疾病的发生发展有相同的作用机制,对这些变异的鉴定将促进我们对AIDs 的进一步认识,为今后的研究提供新的思路。

表1 常见AIDs 中的外显子变异情况Table 1 Exon variants in common AIDs
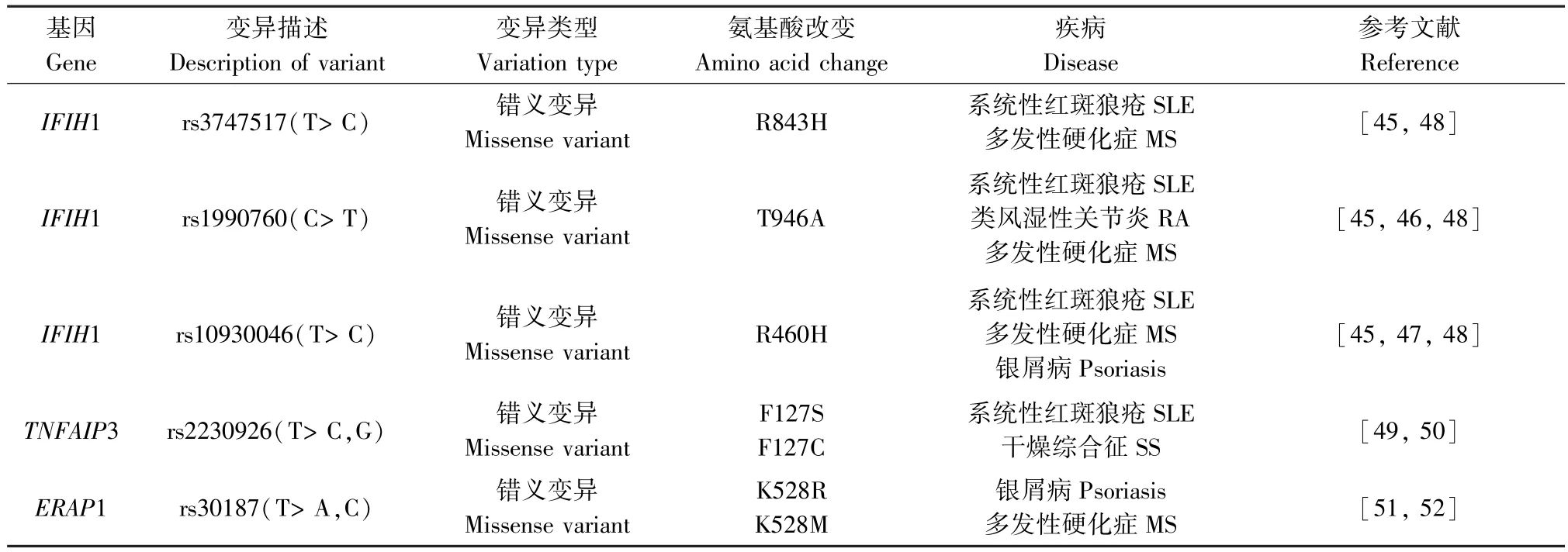
表2 不同AIDs 共性的外显子变异情况Table 2 Common exon variants involved in different AIDs
7 小结与展望
综上所述,WES 在多种AIDs 中的使用已新鉴定了多个疾病独有或共有的遗传变异,这些变异位点通过各种不同的途径参与疾病的发生发展,对AIDs 的发生发展提出了新机制。 但是,这些相关联的遗传变异如何参与AIDs 发生发展的具体分子机制,尚未完全阐明,有待于进一步的研究。 随着测序技术的进步及广泛应用,WES 将来可能有望与蛋白组学、表观遗传学等多组学联合测序,辅以细胞成像技术、原位杂交、免疫组织化学等技术,促进对复杂疾病中基因异常表达调控机制及后续修饰的认识,为AIDs 的预防、早期诊断、监测、治疗及临床预后提供重要的参考。
