离子键型石墨层间化合物及其应用于碱金属离子电池的研究进展 *
2021-07-12柳小玄刘洪波刘金水
柳小玄,李 铮,韩 飞,刘洪波,刘金水
(湖南大学 材料科学与工程学院 先进炭材料及应用技术湖南省重点实验室,长沙 410082)
0 引 言
石墨中碳层与碳层之间依靠弱的范德华力结合(碳层内σ键键能345 kJ·mol-1,层间范德华力16.7 kJ·mol-1[1]),这使得多种分子、原子、离子和原子团能进入石墨层间形成不破坏石墨层状结构的新化合物,称为石墨层间化合物(Graphite Intercalation Compounds,GICs)[2-4]。根据客体与宿主石墨之间形成的化学键类型,广义上将GICs分为共价键型GICs和离子键型GICs[5-6]。共价键型GICs中碳层的碳原子经过sp3杂化使得碳层结构弯曲变形,成为无自由电子的绝缘体,产物种类较少,因此“GICs”一词多数情况下都指离子键型GICs[7]。电化学体系中化学物质嵌入石墨层间形成离子键型GICs的反应可以表示为:
C+mX→CXm
(1)
石墨和嵌入其中的客体(X)发生氧化还原反应,如果客体(X)成为阴离子(Xm-),那么这个可逆反应可以表示为:
Cx+X→Cxm+·Xm-
(2)
由于客体X接受了碳层中的离域π键的m个电子,所以这类GICs称为受电子型GICs。如果客体X向碳层提供电子,那么这种GICs就称为供电子型GICs,可逆反应表示为:
Cx+X→Cxm-·Xm+
(3)
客体物质进入石墨层间后,减弱了石墨碳层中离域π电子相互作用,且客体与宿主之间发生电荷转移,增加了材料的载流子(电子或空穴)浓度并在相邻碳层间搭建高效的载流子通道,从而赋予GICs优良的导电性[8-11]。
由于离子键型GICs客体的插入,碳层中的离域π键分别从/向插层客体接受/提供电子,引起石墨碳层的电子特性显著变化并赋予它独特的性质。通过有目的地改变插层客体种类,可以便捷地调整产物结构特征和理化性质,因此离子键型GICs受到越来越多的关注。本文选取碱金属和金属氯化物分别作为供电子型和受电子型GICs的典型客体代表,阐明前者在石墨中的插层行为,综述后者嵌入石墨碳层间之后在碱金属离子(Li+,Na+,K+)电池中的应用,最后对离子键型GICs电极材料未来的发展趋势进行展望,旨在为新一代绿色环保、高功率/能量密度和低成本电极材料的开发提供思路。
1 碱金属-GICs中客体的插层行为
碱金属容易失掉最外层电子,进入石墨碳层后形成供电子型的碱金属-GICs。碱金属-GICs中,随着碱金属离子浓度增加,石墨碳层间距变大,层间范德华力减弱[12-13]。Safran等[14]首次提出,成阶机理与宿主客体之间的长程弹性相互作用和静电相互作用有关。在碱金属-GICs中,电荷从碱金属转移到了石墨,所以c轴方向存在着碳层之间的排斥力,但同时石墨碳层并未剥离,还有层间范德华力维系。碱金属插层石墨过程中,阶结构从高阶到低阶的形成过程伴随着层间引力和斥力的动态变化[15]。
为了更好地理解插层过程,研究者们提出GICs结构模型。图1为客体插层石墨Daumas-Hérold模型(也称阶畴模型),客体在石墨层内引起局部碳层变形,层间有插入物的部分隆起,与周围未有插入物的部分形成褶皱层[16]。在碱金属离子嵌入石墨的过程中,阶结构发生转变。碱金属离子倾向于经过不连续的步骤,周期性地沿垂直c轴的方向进入石墨层。客体嵌入引起的体积膨胀从石墨边缘开始,到中心部位为止。插层反应过程中,客体不断向石墨内部推进,整体结构产生明显形变。接下来分别简述Li,K,Na嵌入石墨的插层行为及储存机理。
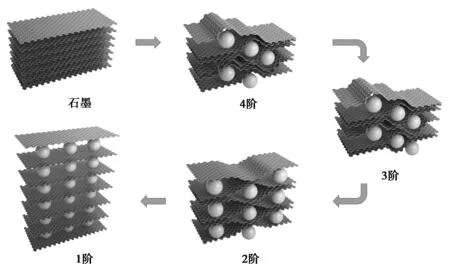
图1 客体插层石墨的Daumas-Hérold模型Fig 1 Schematic illustration of the Daumas-Hérold model of guest-intercalation into graphite
1.1 锂金属-石墨层间化合物(Li-GICs)
锂离子嵌入石墨时,主要在面内方向扩散,X射线散射技术的研究结果表明电荷从Li转移到碳[17]。完全锂化时,得到一阶LiC6,每个锂原子占据六角碳环的中心,处于最低能量状态,所有Li的位置等同(α位)[18],理论容量为372 mA·h·g-1(图2a)。
最近,Moriwake 等[19]发现从Na到Cs,碱金属-GICs的形成能越来越小,这表明从Na到Cs形成的碱金属-GICs越来越稳定,但Li是这个趋势的例外(图2b),因为锂碳键有更多的共价组分(图2c),这使得Li-GICs比其他的碱金属-GICs更稳定。在阶转变过程中,石墨层的堆积顺序发生变化,相邻的碳层直接相对(A A堆叠)。插层时,阶结构变化过程会受到电解液种类、宿主石墨种类和测试条件的影响,但总的阶结构转变方向始终是按照3阶→2阶→1阶的顺序进行[13,20-21]。

图2 (a)LiC6的结构[12],(b)碱金属-GICs的形成能[19],(c)LiC6,NaC6和 KC6的电子密度[19],(d)石墨负极的LIBs工作原理示意图[25]Fig 2 (a) Structure diagram of (b) calculated formation energies of AMC6 for AM = Li, Na, K, Rb, and Cs [19]; (c) electron densities for compositions LiC6, NaC6 and (d) schematic illustration of operating principles of LIBs with anode of graphite [25]
在过去几十年里,研究者们探索了大量碳质材料用作锂离子电池负极的情况,包括一维碳纳米管,碳纳米线,碳纳米纤维[22],二维功能石墨烯纳米片[23-24]。它们具有高比表面积,有利于电解液迅速渗透,离子快速扩散和离子大量存储,但也会引起更多电解质分解,形成过多的固体电解质界面(SEI:solid Electrolyte Interface)层,从而导致初始库伦效率低。石墨具有相对较低的比表面积,较小的体积膨胀,较高的初始库仑效率和丰富的自然资源,所以有充当锂离子电池负极材料的独特优势(图2d)。为了更好地适应当下对储能设备高容量、高倍率充放电的要求,未来需进一步探索锂金属在石墨内部的插层行为及储存机制,便于对石墨进行改性研究,以便获得更为优异的电化学性能。
1.2 钾金属-石墨层间化合物(K-GICs)
关于K-GICs结构和化学/物理性质方面的研究早在20世纪30年代就已经展开,KC8是第一种被发现的碱金属-GICs,可以通过将石墨浸润在钾蒸汽或排水性含钾溶液中合成[26-27]。上世纪90年代,Marassi等[28]用KC8作锂离子电池的负极,发现在充电过程中K+成功地从KC8中脱出。2010年,Yang等[29]发现在1 163 K高温条件下KF的熔融盐中,钾能够可逆嵌入石墨,但得到的K-GICs材料热稳定性差,在空气中迅速分解,这严重阻碍石墨作钾离子电池负极的发展,因此对钾离子电池的开发提出了巨大的挑战[30]。然而,2015年Ji等[20]首次报道采用非石墨类软碳用作钾离子电池负极,证实钾化过程中依次形成KC36、KC24和KC8并表现出273 mA·h·g-1的高可逆容量,自此钾离子电池用石墨电极吸引了众多目光。
如图3(a,b)所示,在钾插层过程中,初期第1-3点对应的三条曲线内石墨的衍射峰逐渐减弱,第4点所对应的曲线出现KC36(3阶K-GICs,见图3c)在22.0°和29.4°处的特征峰。进一步钾化过程中,第5点XRD曲线显示,KC36转变成KC24(2阶K-GICs),在20.2°和30.6°出现新特征峰。第6、7点对应的曲线中经历两相共存阶段,第8点对应曲线位于16.4°和33.4°处特征峰表明KC8(一阶K-GICs)形成。恒电流充电过程中相转变与放电过程相反,这些标志性阶结构的出现和消失,表明阶结构转变具有可逆性。在K+的嵌入过程,石墨层间距从0.335 nm(初始石墨)增加到0.535 nm(KC8),体积膨胀了61%,而一个完整的恒电流钾化/去钾化循环可以从石墨到KC8再回到石墨。这表明K插层带来的巨大体积改变没有破坏石墨原有的层状结构,意味着石墨碳层结构可以产生较小的应变来实现对碱金属的可逆储存和释放[31-32]。

图3 K-GICs [20](a)恒流钾化/去钾化电压曲线,(b)对应a图标记状态的XRD曲线,(c)不同阶数K-GICs的结构示意图Fig 3 K-GICs[20]: (a) First-cycle galvanostatic potassiation/depotassiation potential profiles of K-GICs; (b) XRD patterns of electrodes corresponding to the marked selected charge states in (a); (c) structure diagrams of different K-GICs
单一客体K+插层石墨的反应电位约0.2 V(vs. K/K+),高于K的沉积电位,较大的电压差能够有效地抑制钾枝晶的形成,从而提升负极材料的循环稳定性能,但另一方面也意味着全电池的输出电压低,造成电池能量密度较低等问题[13,20]。然而溶剂化离子插层使得石墨通常有很好的循环稳定性能。Gao等[33]发现,K+在碳酸乙烯酯/碳酸二甲酯电解液中插层石墨,石墨表面形成疏松的SEI膜,体积膨胀63%;但采用二甲醚(DME)电解液时,K+插层石墨后石墨表面依然光滑,没有明显的SEI膜生成,体积膨胀率仅7.7%。这归因于DME对K+的配位数更大,由此带来溶剂化的键合更强,且[K-DME]+的最低未占分子轨道比石墨的更高,因此不与石墨反应生成SEI膜。虽然循环稳定性有提升,但溶剂参与的共插层反应也会带来体积能量密度低和比容量低等问题,K-GICs在钾离子电池(KIBs)中的应用仍需继续探索。
1.3 钠金属-石墨层间化合物(Na-GICs)
早在1985年研究者们就从电子结构的角度发现Na与C之间关联微弱[34]。2013年,Nobuhara等[35]计算碱金属-GICs的形成能,证实了Na-GICs在热力学上是不稳定的化合物,其能量不稳定的原因是由于Na插层后石墨中碳碳单键的键长大幅改变。2014年,Grande等[12]修改平面内碳碳单键的键长模型发现,与LiC6、KC6相比,NaC6中碳碳单键并不稳定(图4a)。范德华密度函数理论(vdW-DF)研究结果表明,Na与石墨反应所生成NaC6和NaC8的生成焓大于零,意味着这两种产物在热力学上难以稳定存在[12]。2016年,Goddard等[36]通过量子力学计算发现,由于碱金属原子电离和碱金属客体-宿主耦合之间存在竞争,所以Na在同族碱金属中与宿主石墨的化学键结合最弱。但是,这种限制似乎能通过将Na+溶剂化来解决在石墨碳层间的插入问题。
通过选择合适的溶剂使Na+溶剂化,在溶剂化外壳的作用下,Na+插入到石墨碳层空间,形成稳定的化合物。必须指出的是,溶剂与石墨层或碱金属离子之间的相互作用,对共插层进程有决定性影响。从2014年Adelhelm等[37]开始,兴起了使用二甘醇二甲醚对Na+溶剂化,从而实现Na+嵌入石墨碳层间的研究热潮[38-40]。为了更好地对Na+进行溶剂化作用,学者们从醚基电解液方面进行了研究,包括单甘醇二甲醚、二甘醇二甲醚、三甘醇二甲醚、四甘醇二甲醚等(图4b)。
2015年,Kang等[42-43]首先发现,在上述不同链长的线性醚类中,链更长的分子溶剂化Na+的能力更强,这是由于Na+之间相互排斥而长链溶剂与Na+配位后,溶剂链越长则配位后相邻Na+间距越大,由此带来Na+之间斥力显著降低。2019年,Kang等[44]发现,醚类溶剂的链长对Na+-溶剂共插层石墨的电位有影响,溶剂链越长,共插层电位越高。使用长链溶剂时,溶剂化复合离子的体积更大,因而产生了较大层间距,就使得石墨碳层之间的排斥力减小,从而产生更高的插层电位。Han等[39]通过DFT计算表明,二甘醇二甲醚与石墨碳层之间的范德华相互作用保持了三元GICs的结构完整性。有研究发现,扁平的二甘醇二甲醚分子能够完全溶剂化Na+,这可以加速Na+-二甘醇二甲醚在石墨层间的扩散(图4c)。Kang等[43]根据重量变化测试和能量色散X射线光谱分析得出结论,共插层时二甘醇二甲醚分子和Na+的比例是1∶1。然而,Yu等[38]使用从头计算法(ab initio method),从碳层和二甘醇二甲醚之间离子键的强度方面考虑,发现共嵌结构中两个二甘醇二甲醚分子包围一个Na+比一个二甘醇二甲醚分子包围一个Na+所形成化合物的结构更为稳定。2016年,Gotoh等[40]通过化学实验合成了Na-diglyme-GICs,又通过元素分析测量出每两个二甘醇二甲醚分子与一个Na+配位。此外,固态2H 核磁共振谱(NMR)结果表明,二甘醇二甲醚分子通过一个氧原子与Na+微弱地配位(图4d),并绕O-Na轴旋转,二甘醇二甲醚由此运动,以加速共扩散。应当注意,二甘醇二甲醚和Na+的配位数与配位结构仍然存在争议,未来还需更详细的理论研究和实验证据[37-38,45]。

图4 (a)碱金属-GICs的平面内碳碳单键长键(L)短键(S)[12],(b)Na+与线性醚类复合物的球棍模型[41],(c)使用二甘醇二甲醚溶剂时石墨储钠示意图[42],(d)二甘醇二甲醚和钠在石墨中的模型[40]Fig 4 (a) In-plane C-C long (L) and short (S) bonds in alkali metal intercalation compounds [12]; (b) ball-and-stick models of Na+(Gx)y complexes [41]; (c) proposed schematic of Na storage in natural graphite using DEGDME [42]; (d) models of diglyme-solvated sodium in graphite [40]
总之,柔韧线性醚类溶剂虽然高压稳定性较差[46-47],却有良好的配位作用,作石墨电极的电解液溶剂时能够促使Na+插入到石墨碳层内部形成GICs,从而表现出最令人满意的电化学性能。基于这种溶剂依赖性,将不同溶剂混合被视为调控共插层行为的有效途径[48]。寻找合适的溶剂能帮助Na/石墨电池可逆地存储和释放溶剂化Na+,从而实现共嵌插层反应,这也为LIBs甚至是KIBs的共嵌应用奠定了研究基础。
2 金属氯化物-GICs的结构设计与电化学性能
金属氯化物-GICs作为受电子型GICs的杰出代表,在客体与碳层相互作用的条件下产生了一系列新特性,如高导电、高导磁、超导、吸波、催化等[49-52],吸引了众多学者进行理论和实验研究。FeCl3、MnCl2、AlCl3、CuCl2等固态金属氯化物具有层状或链状结构,金属阳离子位于Cl构成的八面体中心,每个阳离子被4或6个阴离子包围,每个Cl被2个或3个阳离子共享。这样的平面层状或链状结构使金属氯化物微粒运动受限,因此所制备的MClx-GICs具有很高的热力学稳定性[53]。接下来,本文将从FeCl3-GICs储锂储钾机制出发,讨论FeCl3-GICs材料的修饰改性对其电化学性能的提高,同时探讨其他金属氯化物插层的GICs的电化学储能特性。
2.1 FeCl3-GICs的储锂机制
FeCl3插层反应操作简便、产物结构稳定,是目前金属氯化物-GICs领域最常用、研究最深入的插入剂[54]。Xia等[55]通过融盐法使FeCl3进入石墨层间,形成FeCl3-GICs,如图5(a)。将其用在锂离子半电池中发现,400次循环后,FeCl3-GICs的容量保持率为100%,可逆容量高达500 mA·h·g-1。
为了探明FeCl3-GICs的储锂机制,Xia等[55]采用XRD表征半电池中不同充放电程度的FeCl3-GICs电极,如图5(b,c)。当电池放电,电压从3 V降到0.75 V(vs. Li/Li+)时,FeCl3-GICs的特征峰基本消失,对应着FeCl3的分解,表明有相转变反应发生。作者推断转化反应为FeCl3+ 3Li++ 3e-→Fe+3LiCl,且产物Fe和LiCl是纳米尺寸,颗粒太小以至于X射线无法检测到。这种推测在之后得到验证,该材料被继续放电到0.01 V,放电后电极用高分辨透射电子显微镜(HRTEM)分析,发现确有3~6 nm的Fe颗粒均匀分布在石墨层中[56]。图5(b,c)中,当Li/FeCl3-GIC纽扣电池持续放电,电压下降到0.005 V时,G(002)从26°移到24~25°,对应Li+进入石墨层间形成1阶和2阶Li-GICs的过程。充电至电压达到3 V时,XRD曲线中没有出现任何FeCl3-GICs的特征峰,G(002)又回到26°,这表明Li+从石墨层中脱出,第一次放电后FeCl3-GICs原本的结构不能够可逆恢复,产物为纳米级FeCl3夹在石墨层间所形成的复合物。

图5 FeCl3-GICs表征[55]:(a)FeCl3-GICs的结构和锂化反应机制示意图,(b)初始充放电曲线,(c)不同充放电状态下的XRD曲线Fig 5 Characterization of FeCl3-GICs [55]: (a) schematic illustration of the structure and the Li intercalated mechanism; (b) initial discharge-charge profile of FeCl3-GIC; (c) ex-situ XRD patterns of the FeCl3-GIC electrode at different discharge/charge states in the initial cycle
上述反应机制(图5a)可以解释FeCl3-GICs在储锂时所表现出的高容量,其来自:(1)锂离子在石墨层间嵌入/脱出;(2)锂离子在石墨碳层表面吸附/脱附;(3)LiCl的合成/分解。之后,Qian等[56]通过选择膨胀石墨做宿主材料合成FeCl3-GICs,作为锂离子存储材料时可以有效提升可逆容量,容量达到719 mA·h·g-1。FeCl3-GICs的层状结构和高电子导电性使其作为锂离子电池负极材料表示出良好的电化学性能,这为金属氯化物-GICs在电化学储能电池领域的应用研究拉开了帷幕。
2.2 FeCl3-GICs的储钾机制
因为钾储量丰富,分布广泛,所以钾离子电池在取代锂离子电池方面被寄以厚望[57-58]。可是K+半径较大(K+: 0.138 nm,Li+: 0.076 nm)[20,32,59],在石墨中扩散速率低,导致石墨负极在钾离子电池中的倍率性能非常差。为了克服这种弊端,现已广泛研究了各种碳质材料,例如掺杂的石墨烯[60],活性碳[61],硬碳[62]和碳纳米纤维[63],但仍未取得令人满意的结果。然而,GICs具有优异的导电性和扩展的层间距,在钾离子存储方面极具应用潜力。
2018年,Ci等[64]用两步法制备出FeCl3插层膨胀石墨(FeCl3-EG),并用于钾离子电池负极,如图6(a),所得FeCl3-EG比表面积高达85.23 m2·g-1,约为石墨(10.58 m2·g-1)的八倍。其中微孔与介/大孔共存,数量众多,能提供丰富的电极-电解质界面和离子电子运输通道。同时,高比表面积和孔体积可以缓冲K+嵌入/脱出带来的体积改变。作者用XRD探究其储钾原理(图6b,c),FeCl3-EG的1(001)和1(002)的特征峰在电极放电的过程中逐渐消失,之后又伴随充电过程逐渐显现,但恢复程度比初始状态要弱,这是因为第一次放/充电过程中K+嵌入/脱出引起石墨一定程度的晶格畸变。另一方面,1(003)的特征峰在整个充放电过程一直存在,表明FeCl3-EG的层状结构稳定存在,能为K+嵌入/脱出提供层状骨架支持。FeCl3-EG电极完全放电后,在高分辨透射电镜下能观察到有粒径5~8 nm的Fe颗粒均匀分布在石墨层间,所以证明有FeCl3+3K++3e-→Fe+3KCl反应发生[64]。
将K/FeCl3-EG纽扣电池在100 mA·g-1的电流密度下测试,在第一次循环中表现出放电容量762 mA·h·g-1,充电容量260 mA·h·g-1,库伦效率约34%。这种初始不可逆容量来自第一次放电过程中电解液分解,在FeCl3-EG电极表面生成SEI膜,和第一次充电时部分KCl不可逆分解[64]。当电流密度从50增加到5000 mA·g-1时,FeCl3-EG展示出令人惊叹的倍率性能(图6d)。在电流密度50 mA·g-1时,放电容量为269.5mA·h·g-1,然而在电流密度高达5 000 mA·g-1的情况下,放电容量仍保持133.1 mA·h·g-1,当电流密度再降到50 mA·g-1时,容量又恢复到266.5 mA·h·g-1[64]。与此产生强烈对比的是,未经插层处理的石墨在KIBs中,电流密度5 000 mA·g-1的条件下,仅有容量约18.1 mA·h·g-1。因此,FeCl3-EG的储钾性能明显优于商业石墨电极,是钾离子电池领域富有前景的负极材料。

图6 FeCl3-EG [64]:(a)合成过程和充放电行为,(b)初始充放电曲线,(c)不同充放电程度的FeCl3-EG电极异位XRD图谱,(d)石墨和FeCl3-EG电极在不同电流密度下的倍率容量Fig 6 FeCl3-EG [64]: (a) illustration of the synthesis process and the charge/discharge behavior; (b) initial discharge-charge curves; (c) ex-situ XRD patterns of the FeCl3-EG electrode at different discharge-charge depths; (d) rate capability of graphite and FeCl3-EG electrodes at various current densities
2.3 FeCl3-GICs的结构设计及储能应用
FeCl3-GIC在锂离子电池中的使用仍处于探索阶段,因为插层剂FeCl3吸水性强,导电性差,易溶解到溶剂中,带来穿梭效应而引起严重的容量衰减[65],所以需要设计可行有效的FeCl3插层结构,增强其作为负极材料的循环稳定性。Wang等[66]在惰性气体保护下,670 ℃高温处理2阶FeCl3-GIC,得到FeCl2纳米晶体夹在Cl掺杂的石墨层中(C-Cl / FeCl2/C-Cl)的复合物,在锂离子存储方面表现出优异的倍率性能和长循环寿命。随后,Yu等[67]进一步制备出FeCl3插层少层石墨烯(FeCl3-FLG),展示出高可逆容量。但是,放电产物LiCl的粒径比铁颗粒大得多[68-69],因此锂插层过程中石墨层间距将进一步扩大,这又会加剧氯化物在电解质中的溶解和在正负极之间的穿梭,导致FeCl3-GIC电极材料在充放电过程中容量衰减和库伦效率降低,所以难以获得令人满意的GICs负极。
针对离子键型GICs中金属氯化物客体的溶解和穿梭问题,本课题组提出了三种有效的解决策略:(1)引入极性锚定位点,(2)筛选石墨宿主结构,(3)设计插层客体种类。
在第一种策略中,由于插入客体金属氯化物是极性物质,根据“极性-极性相互作用”原理,在石墨层间化合物内部引入极性位点用于锚定金属氯化物,从而抑制其溶解和穿梭,极大程度提升GICs负极的循环稳定性能。第一性原理计算表明,FeCl3与环氧官能团修饰的石墨层之间的吸附能明显高于FeCl3与原始石墨之间的吸附能。为此,本课题组Zhang等[70]通过在石墨碳层上引入丰富的环氧官能团,利用强大的化学键合将氯化物固定在含环氧基团的石墨层间空间内 (图7a)。这样制备出的FeCl3-HOGIC(High Oxidized Graphite Intercalation Compound)负极经历50个循环后,可逆容量1371 mA·h·g-1,容量保持率达98%(图7c)。对完全锂化后的FeCl3-HOGIC电极进行XRD分析,发现产物为LiC,而不是传统的LiC6。石墨储存锂离子的反应机制变为C + e-+Li+→LiC(图7e),由此带来锂离子存储容量大幅度升高。具有丰富含环氧基团的FeCl3-HOGIC是锂离子电池用高容量、高稳定性的负极材料,这一发现将极大提升石墨类材料在锂离子电池中的应用潜力,有力推动高能量密度锂离子电池的研发。

图7 (a) FeCl3与不同宿主结合的结构模型图和键能,G代表石墨层,OG代表含环氧基团的石墨层 [70],(b) FeCl3与石墨(G)和Fe2O3结合的结构模型图及键能 [71],(c) FeCl3-HOGIC、FeCl3-RHOGIC、FeCl3-LOGIC和FeCl3-GIC的循环性能比较 [70],(d)不同辐照时间处理的Fe2O3/GIC和FeCl3-GIC循环性能的比较 [71],(e)FeCl3-HOGIC储锂机制示意图 [70],(f) Fe2O3/GIC(上)和FeCl3-GIC(下)电极循环后结构变化示意图 [71]Fig 7 (a) Structure models and simulated adsorption energies between the FeCl3 guest and different graphite hosts: graphite layer(G) and graphite with epoxy functional groups(OG) [70]; (b) structural models and calculated binding energies for FeCl3 with graphite layer(G) and (c) comparison of the cycle performance of FeCl3-HOGIC, FeCl3-RHOGIC, FeCl3-LOGIC and FeCl3-GIC [70]; (d) comparison of cycle performance of GIC and Fe2O3/GIC [71]; (e) schematic illustration of the FeCl3-HOGIC with the lithium storage mechanism [70]; (f) schematic illustration of the structural change in Fe2O3/GIC and FeCl3-GIC electrodes after cycles [71]
为了进一步提升极性位点的化学锚定作用,本课题组Li[71]等采用更为便捷的微波辅助氧化法,在FeCl3-GICs的石墨层间边缘引入适量的碳层状极性物质Fe2O3,制备出Fe2O3/GIC。利用丰富的极性表面限制金属氯化物的向外扩散,从而有效抑制FeCl3在循环过程中的溶解(图7b)。充放电过程中Fe2O3/GIC电极电导率增加,活性位点逐渐展开,显著提高了电池的循环稳定性(图7d)。图7(f)展示了FeCl3-GIC和Fe2O3/GIC在电化学循环过程中的结构变化:FeCl3-GIC中由于石墨层和氯化物之间作用微弱,不能阻止FeCl3溶解和穿梭,所以电极的活性组分损失严重;层状Fe2O3/GIC中有丰富的活性位点,通过极性作用固定氯化物[72-73],提高循环稳定性。该工作不仅利用简单的内转化法解决了金属氯化物溶解穿梭的技术难题,同时也加深了人们对石墨层间化合物材料的形貌、结构和性能之间的相互关系的认识,为进一步设计电化学储能用石墨层间化合物材料提供了新思路和新途径。
第二种解决策略是通过筛选具有独特微观结构的石墨宿主用于负载金属氯化物,利用微观结构的限制作用来阻止金属氯化物的溶出,从而提高GICs的循环稳定性。金属氯化物-GICs常见的石墨宿主有天然鳞片石墨,微晶石墨,膨胀石墨,氧化石墨和多层石墨烯等。Xia等[55]制备的FeCl3-GIC循环比容量为480 mA·h·g-1。后来,本课题组Sun[74]等选取具有较小的微晶尺寸、扭曲的微晶碳层和各向同性结构的微晶石墨作宿主,制备FeCl3-MGIC(Microcrystalline Graphite Intercalation Compound)。利用石墨扭曲的碳层和各向同性结构抑制金属氯化物的溶解穿梭问题,所得到的FeCl3-MGIC在100个循环周期内可逆比容量稳定在905 mA·h·g-1,有效地提升了该类负极材料的循环性能,极具应用前景。此项工作不仅揭示了石墨宿主结构对石墨层间化物的生成及电化学性能的影响规律,也为微晶石墨资源高附加值的利用提供了有效参考。
2.4 其他金属氯化物插层及其碱金属离子存储应用
金属氯化物插层石墨的客体种类丰富,很早就有FeCl3,MoCl5,BiCl3,CuCl2,TaCl5,ZnCl2,SnCl2,CdCl2,NiCl2等插层石墨,相应的反应温度、时间和石墨/插入物的摩尔比列入表1中。为了应对金属氯化物客体的溶解和穿梭问题,本课题组提出的第三种策略是设计插层客体种类。由于不同金属氯化物客体与石墨宿主之间的吸附能不同,为了提升两者之间的作用力,2020年本课题组Li等[78]设计出具有纯一阶和二阶结构的MoCl5-GIC,首次报道了MoCl5-GIC用作高体积能量密度的钠离子电池负极材料。
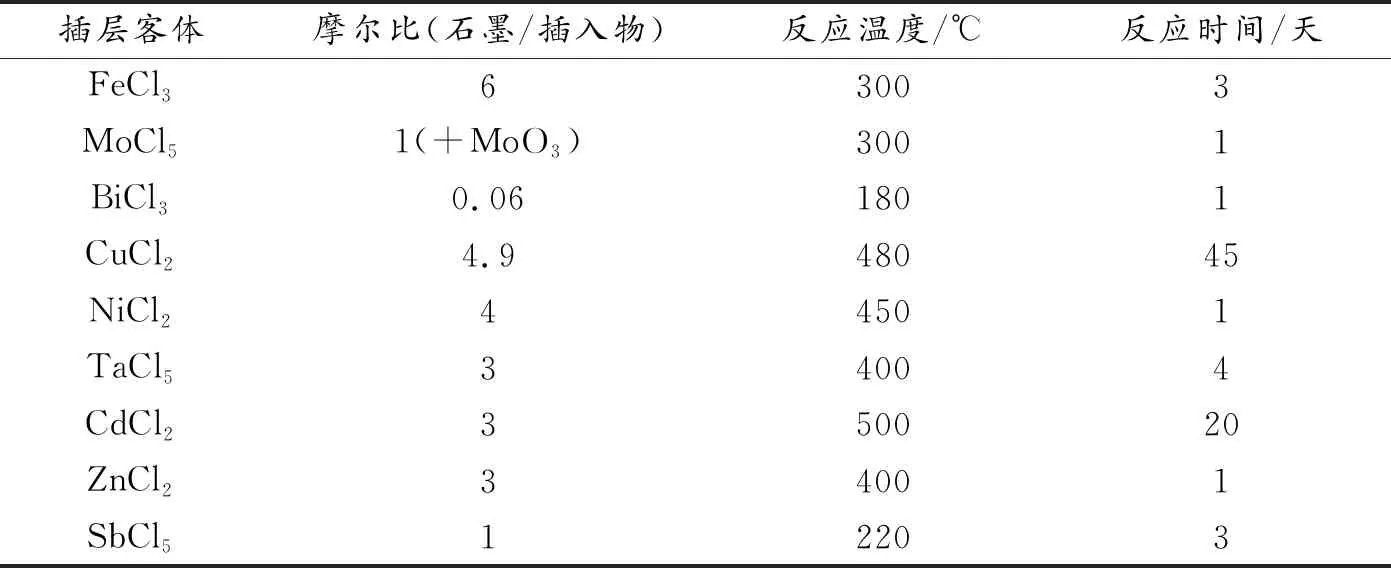
表1 制备金属氯化物-石墨层间化合物的反应条件[68-69,75-77]
第一性原理计算表明MoCl5和石墨之间的吸附能(图8a,Eb=0.21542 eV)明显高于FeCl3和石墨之间的吸附能(图7a,Eb=0.05875 eV),意味着MoCl5能有效插入并紧密固定在石墨碳层中。1阶MoCl5-GIC具有平行对齐的晶格条纹,石墨碳层间距扩展到0.922 nm(图8b),但小于FeCl3-GIC的层间距(0.938 nm),MoCl5-GIC中相对小的层间距既为钠离子存储提供充足的空间,又使得插入的MoCl5具有优异的热力学稳定性。将MoCl5-GIC用作钠离子半电池负极,通过XRD表征MoCl5-GIC电极完全嵌钠后的产物(图8c),发现存在NaCl,NaC64,Na-GIC物质。其中,Na-GIC的存在表明有少部分二甘醇二甲醚溶剂分子与钠离子发生溶剂共嵌,进入石墨层间。结合循环伏安曲线推断储存钠离子时发生转化反应MoCl5+5Na++5e-→5NaCl+Mo。MoCl5-GIC结构致密,体积密度大,由此带来在钠离子电池中体积容量高达426 mA·h·cm-3,有利于小型电池高能量密度化发展,储钠时最大比容量为275 mA·h·g-1,电流密度0.1 A. g-1条件下循环100次后容量保持率91.4%(图8d)。当电流密度1 A. g-1时,MoCl5-GIC电极储钠容量为255 mA·h·g-1,容量保持率为81.4%,容量衰减率极低,平均每次循环仅0.018%。MoCl5与石墨层之间强烈的相互作用有助于在反复的充/放电过程中,固定可溶性氯化物,从而产生优异的钠离子存储循环性能,为石墨类材料在钠离子电池中的应用提供了行之有效的方法。

图8 MoCl5-GIC [78]:(a) MoCl5/Graphite的结构模型和计算结合能,(b) MoCl5-GIC的高分辨透射电子显微镜图像,(c)MoCl5-GIC电极在完全钠化后的XRD图谱,(d)MoCl5-GIC和FeCl3-GIC循环性能的比较Fig 8 MoCl5-GIC [78]: (a) the structural models and calculated binding energies of MoCl5/graphite; (b) TEM images of MoCl5-GIC; (c) XRD pattern of the MoCl5-GIC electrode under full sodiation conditions; (d) comparison of the cycle performance of MoCl5-GIC and FeCl3-GIC
此外,SbCl5-GIC、NbCl5-GIC和TaCl5-GIC在空气中难以稳定存在,目前的研究集中在探究不同种类的宿主石墨对其在水中和空气中稳定性的影响。FeCl3-NiCl2-GIC的探索聚焦在不同制备方法对产物催化还原和微波吸收性能的影响。FeCl3-AlCl3-GIC的关注方向在不同宿主对产物的导电性和空气稳定性的影响。这些离子键型GICs在碱金属离子存储方面存在研究空白,有很大的研究空间亟待探索。
3 结 语
离子键型GICs具有独特的物理化学性质和优良的电化学行为,在电化学能量存储领域扮演着越来越重要的角色。全球电池技术迅速发展,对未来离子键型GICs的开发提出了新的要求,尤其在如下五个方面还有很多工作值得展开:(1)在溶剂分子的帮助下可促进碱金属离子在石墨内部的插层,用于设计制备电解质溶剂和碱金属离子共插层的三元GICs材料,丰富离子型GICs的种类和性质;(2)石墨内部多种金属氯化物共同插层,多元客体之间彼此相互作用对化合物的拓扑形态、电子结构以及插层行为的影响以及对碱金属离子存储性能的影响尚不明确,未来需加强这方面的研究工作;(3)石墨晶体结构多样,石墨宿主的物理化学特性对GICs的形成以及离子存储方面的电化学性能影响尚未清楚,亟需开展这方面的研究工作;(4)插层客体的选取无须局限在氯化物之内,未来可以通过不同的转化手段将氯化物客体转化为硫化物、氧化物等,客体种类的变化又会赋予GICs存储碱金属离子的新机制;(5)随着金属离子电池的蓬勃发展,离子键型GICs能够储存的金属离子种类将不再局限于碱金属离子,可以扩展至存储能量密度更高的Mg2+、Zn2+、Al3+等。
