GPR30激活后下调P53-PUMA信号抑制全脑缺血诱导的海马神经元线粒体凋亡途径
2021-07-12郭思含孙立萍赵雨逢王瑞敏胡书群
郭思含,孙立萍,赵雨逢,韩 东,王瑞敏,胡书群
0 引 言
全脑缺血导致的认知功能障碍是心脏骤停(cardiac arrest,CA)生存者最普遍的后遗症。大鼠四动脉结扎全脑缺血(global cerebral ischemia,GCI)模型可模拟心脏骤停诱发的海马CA1区神经元迟发性死亡[1]。目前,亚低温治疗是治疗CA的唯一措施,进一步研究发现温度降低时冷诱导RNA结合蛋白表达显著增强,可有效保护CA大鼠海马CA1区神经元损伤[2]。然而,Meta分析显示,亚低温治疗并不能有效改善CA幸存者认知功能[3],亟待探求其所致神经元损伤的机制和有效的治疗措施。
近期,雌激素膜受体--G蛋白耦联雌激素受体30(G Protein-coupled Estrogen Receptor 30,GPER/GPR30)的神经保护作用受到了研究者关注。GPR30特异激动剂G1具有类雌激素活性。Zhang等[4]研究发现,皮下埋置60 μg/d G1缓释泵30 d可显著降低脑卒中小鼠脑梗死体积,改善中风诱导的外围免疫抑制,并且未发现G1引起血压、血气、血糖等生理指标变化,也未发现引起血清雌激素水平升高、子宫肥大、乳腺细胞增殖等副作用。G36是最新发现的GPR30特异拮抗剂,在结构上G36与G1极其相似;与较早发现的GPR30拮抗剂G15相比,G36对GPR30具有更高的选择性,且与雌激素受体(estrogen receptor, ER)α/β具有更低的亲和力,同时G36可降低雌激素应答元件(estrogen response elements,EREs)的激活[5]。最近,采用GPR30特异激动剂、拮抗剂作为工具药,证实了GPR30激活具有抗神经炎症和抗凋亡作用[6-8]。然而,其具体机制尚不清楚。
P53是一种肿瘤抑制蛋白和转录因子,同时P53也可作为一种凋亡调控蛋白,对诱导细胞凋亡起重要作用。在正常细胞中,P53被一系列调节因子维持在较低水平。其中,小鼠双微体2(murine double minute 2,MDM2)基因作为P53的泛素连接酶,通过泛素化作用促进P53降解[9]。但是,在多种应激刺激如缺血/缺氧、DNA损伤、氧化应激等情况下,P53蛋白被磷酸化激活,从而阻断P53与MDM2的结合和降解,导致P53稳定性增强[10-11]。关于P53促凋亡作用的分子机制,目前有研究认为一是P53转录依赖的细胞核机制,通过促进P21等靶基因蛋白表达促进凋亡;另一是非转录依赖的线粒体机制,即P53直接转位出核,促进线粒体凋亡途径[12]。P53上调凋亡调节因子(P53 upregulated modulator of apoptosis,PUMA)是P53的一个重要靶基因,属于含有BH3结构域的Bcl2家族成员,可通过P53的转录依赖和非转录依赖途径促进细胞凋亡[13]。
在小鼠脊髓损伤模型,GPR30激活可增加抗凋亡蛋白Bcl2水平、下调促凋亡Bax蛋白水平,从而阻断线粒体凋亡途径[14-15]。然而,在大鼠GCI模型,GPR30激活的抗凋亡作用是否与P53信号介导的线粒体凋亡通路有关,尚未见报道。
1 材料与方法
1.1 材料实验动物及主要试剂SPF级成年雌性SD大鼠,3~4月龄,体重250~280 g,购自北京华阜康生物科技股份有限公司,试验动物合格证号:SCXK(京)2014-0004。所有实验动物均在SPF级饲养环境下饲养,自由饮食,室温22~24 ℃。
抗PUMAα/β(sc-28226)、抗Bcl2(sc-7382)、抗GAPDH(sc-32233)一抗购自Santa Cruz公司;抗p-P53(Ser15, ab1431)、抗P53(Ab26)一抗购自abcam公司;抗NeuN(MAB377)一抗购自Millipore公司;抗actin(A2066)一抗购自Sigma-Aldrich公司;Alexa-Fluor 488/568荧光二抗购自Invitrogen公司。TUNEL检测试剂盒(LOT #1639496,Life technologies)购自Active Motif公司;G1(Cat. No.3577/10)、G36(Cat. No.4759)和 Pifi-α(Cat. No. 1267)购自Tocris公司。
1.2方法
1.2.1 实验动物分组与模型制备研究报道,在GCI模型中,海马CA1区神经元损伤出现在24~72 h[1]。因此,本研究选择缺血再灌注7d时间点观察P53信号、GPR30受体激活对缺血后海马CA1区神经生存的影响;选择再灌注1、3 d时间点揭示其可能的分子机制。 具体分组及实施方案如下:实验分组:所有大鼠禁食12 h,均行双侧卵巢切除术(OVX),并随机分为以下9组:假手术组(Sham组):只暴露双侧椎动脉和双侧颈总动脉,不进行电凝和夹闭;缺血再灌注1、3、7 d组(I/R 1d/3d/7d组):缺血12 min,再灌注1、3、7 d;缺血再灌注1、3 d+G1组(I/R 1d/3d+G1组):G1(10 μg/d)于卵巢切除同时颈部皮下埋植微量释放泵,1周后制备缺血再灌注模型,方法同第②组;缺血再灌注3、7d+ Pifi-α组(I/R3d/7d+ Pifi-α组):Pifi-α在缺血前20 min侧脑室微量注射10 μg,连续给药3 d,缺血再灌注方法同第②组;缺血再灌注7 d+G36+Pifi-α处理组(I/R 7 d+G36+ Pifi-α组):G36给药与第⑤组G1给药进行相同操作,G36剂量为(10 μg/d),Pifi-α与缺血再灌注方法同第⑧组2)GCI模型制备OVX术后7 d制备GCI模型[5]。手术第1天,永久性电凝大鼠椎动脉,同时分离双侧颈总动脉,用手术缝合线做标记。24 h后,再次暴露双侧颈总动脉,用动脉夹夹闭12 min,再灌注1、3和7 d。缺血过程中采用加热垫维持大鼠肛温维持在36.5~37.5 ℃。缺血成功的标志为:大鼠在缺血后30~60 s内失去活动能力,翻正反射消失,双侧瞳孔散大,对光刺激闭睑反射消失,眼球颜色由鲜红色变成灰白色,四肢僵直。符合以上标准的大鼠用于本研究。
1.2.2海马冠状冰冻切片的制备及免疫组化参照本实验室已建立的实验方法[5],分别于所需时间点处死大鼠,快速取脑,分离海马CA1区,一半大脑冻存于液氮中,用于Western blot分析;另一半大脑置于4%的多聚甲醛溶液中固定,4 ℃过夜,次日,将固定好的脑块置于30%的蔗糖溶液中脱水,4 ℃储存,至脱水完全。将脑组织用OCT包埋,迅速置于-80 ℃冰箱中速冻,在-20 ℃制备连续冠状冰冻切片(25 μm),-20 ℃储存脑片、备用。
脑组织冠状切片用0.1 mol/L PBS洗涤10 min×3次,0.4%TritionX-100-PBS打孔2 h,10%驴血清室温封闭1 h。一抗孵育4 ℃过夜;用0.1%TritionX-100-PBS洗涤10 min×3次。加相应的荧光二抗(1∶250),室温避光孵育2h。0.1%Trition-PBS洗涤3次,每次15 min。DAPI封片剂封固。采用Olympus FV1000激光扫描共聚焦显微镜观察并采集图像。
1.2.3TUNEL原位细胞凋亡检测技术缺血/再灌注7 d大鼠的脑冠状冰冻切片用于TUNEL原位细胞凋亡实验。实验步骤如下:冰冻切片用0.1 mol/L PBS洗涤10 min×3次。NeuN免疫组化结束后,参照TUNEL试剂盒说明书,将切片置于反应缓冲液A中孵育10 min。在含有酶溶液的TdT反应混合物中,37 ℃恒温箱孵育1 h。dH2O洗涤5 min。在Click-iT Plus TUNEL反应混合物中,37 ℃恒温箱孵育30 min。0.1%Triton-100-PBS洗涤20 min。甘油-PBS封固,激光扫描共聚焦显微镜(Olympus FV1000)下捕获图像,并使用数字成像软件(FV10-ASW1.5)进行分析。
1.2.4Western blot方法按照本实验室已建立的实验方法[3],制备各组海马CA1区组织匀浆液,采用BCA蛋白浓度测定试剂盒检测样品的蛋白浓度。将配平好的蛋白样品加入5×上样缓冲液处理,沸水浴中煮5 min使蛋白充分变性。采用10% SDS-聚丙烯酰胺凝胶电泳分离目标蛋白,电转到PVDF膜上,3%的牛血清白蛋白中封闭2 h,加入相应一抗,孵育4 ℃过夜,TBST洗膜5 min×3次,孵育相应辣根过氧化物酶(HRP)标记的二抗1 h。TBST洗膜10 min×3次,加入ECL化学发光液,图像采集仪器扫描。

2 结 果
2.1 GPR30特异拮抗剂G36逆转Pifi-α的神经保护作用激光扫描共聚焦显微镜下观察结果显示,I/R 7d组海马CA1区NeuN阳性染色细胞数量较假手术组减少(P<0.01),但TUNEL的阳性细胞数量增多(P<0.01)。与I/R 7d组相比,Pifi-α处理组海马CA1区NeuN阳性细胞数增加(P<0.01),而TUNEL阳性细胞数减少(P<0.01)。与单纯给予Pifi-α组相比,GPR30特异拮抗剂G36联合Pifi-α处理组海马CA1区NeuN阳性细胞数量减少(P<0.01),而TUNEL阳性细胞数增加(P<0.01)。见图1,表1。

a:假手术组;b:I/R 7d组;c:I/R 7d+Pifi-α组;d:I/R 7d+Pifi-α+G36组图1 各组大鼠海马CA1区TUNEL技术(红色)和NeuN免疫荧光染色代表性图Figure 1 Representative photographsof TUNEL analysis(red)and IF staining for NeuN (green)

表1 生存神经元和凋亡细胞数量Table 1 The numbers of survival neurons and TUNEL cells
2.2GPR30特异激活剂G1抑制大鼠全脑缺血后海马CA1区P53磷酸化Western blot结果显示,与假手术组相比,GCI后1、3d海马CA1区P53磷酸化水平均升高(P<0.05)。与缺血/再灌注相应时间点(1、3d)相比,用G1激活GPR30受体降低了缺血/再灌注诱导的P53磷酸化水平(P<0.05),见图2。与假手术组相比,缺血再灌注3d组海马CA1区p-P53免疫荧光强度增强,且与DAPI荧光共定位,而G1处理组类似于假手术组,显示海马CA1区p-P53免疫荧光强度较I/R 3d组降低。见图3。
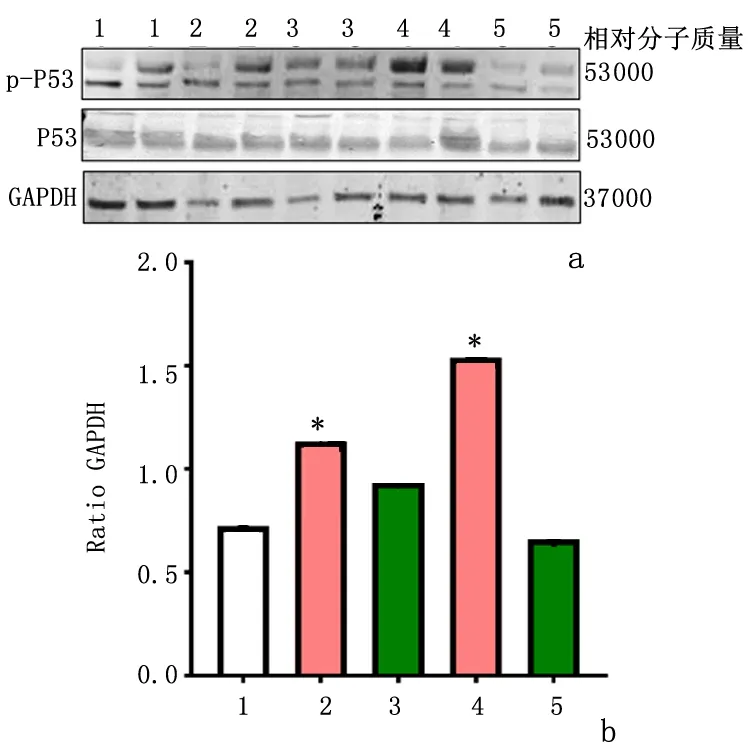
1:假手术组; 2:I/R 1d组; 3:I/R 1d+G1组; 4:I/R 3d组; 5:I/R 3d+G1组a:Western blot检测; b:半定量分析与假手术组比较,*P<0.05图2 GPR30特异激动剂G1降低海马CA1区GCI诱导的P53磷酸化激活Figure 2 GPR30 agonist G1attenuated P53 phosphorylation (activation) induced by GCI in the hippocampal CA1 region

a:假手术组; b:I/R 3d组; c:I/R 3d+G1组DAPI(蓝色)荧光显示细胞核阳性染色图3 各组大鼠海马CA1区p-P53(红色)免疫荧光染色Figure 3 Immunofluorescence staining of p-P53(red)in the hippocampal CA1 region of the rats
2.3GPR30影响P53的初步分子机制研究Western blot结果显示,与假手术组相比,GCI后 1、3d海马CA1区PUMA蛋白水平升高(P<0.05),同时Bcl2蛋白水平降低(P<0.05);给予G1处理抑制了缺血 再灌注诱导的PUMA蛋白水平(P<0.05),反而提高了Bcl2的蛋白水平(P<0.05);并且G1处理组Bcl2蛋白水平高于假手术组对照组(P<0.05)。见图4。与假手术组相比,I/R 3d组海马CA1区PUMA和GFAP荧光强度均增强,且呈现明显的PUMA与GFAP共定位(黄色);而给予G1处理PUMA与GFAP的免疫 荧光强度均较I/R 3d 组降低。Bcl2的免疫荧光强度与PUMA的变化相反,显示I/R 3d组海马CA1区Bcl2荧光强度较假手术组降低,而G1处理升高了Bcl2的免疫荧光强度。见图5。

1:假手术组; 2:I/R 3d组; 3:I/R 3d+Pifi-α组a:Western blot检测;b:PUMA蛋白半定量分析;c:Bcl2蛋白半定量分析与假手术组比较,*P<0.05; 与I/R 3d组比较,#P<0.05图4 P53特异抑制剂Pifi-α对GCI后海马CA1区PUMA和Bcl2蛋白水平的影响Figure 4 Effects of P53 specific inhibitorPifi-α on protein levelsof PUMA and Bcl2 in the hippocampus CA1 region after GCI
2.4GPR30激活减少大鼠全脑缺血后海马CA1区PUMA的蛋白水平、增加Bcl2蛋白水平Western blot结果显示,与假手术组相比,GCI后1、3d海马CA1区PUMA蛋白水平升高(P<0.05),同时Bcl2蛋白水平降低(P<0.05);给予G1处理抑制了缺血再灌注诱导的PUMA蛋白水平(P<0.05),反而提高了Bcl2的蛋白水平(P<0.05);并且G1处理组Bcl2蛋白水平高于假手术组对照组(P<0.05)。免疫组化结果显示:与假手术组相比,I/R 3d组海马CA1区PUMA和GFAP荧光强度均增强,且呈现明显的PUMA与GFAP共定位(黄色);而给予G1处理PUMA与GFAP的免疫荧光强度均较I/R 3d组降低。Bcl2的免疫荧光强度与PUMA的变化相反,显示I/R 3d组海马CA1区Bcl2荧光强度较假手术组降低,而G1处理升高了Bcl2的免疫荧光强度。见图6。

1:假手术组; 2:I/R 1d组; 3:I/R 1d+G1组; 4:I/R 3d组; 5:I/R 3d+G1组a:Western blot检测; b:GAPDH半定量分析; c:Bcl2半定量分析与假手术组比较,*P<0.05; 与I/R 1d组比较,#P<0.05; 与I/R 3d组比较,△P<0.05图5 GPR30特异激动剂G1对GCI后海马CA1区PUMA、Bcl2蛋白水平的影响Figure 5 Effects of GPR30 agonist G1 on PUMA and Bcl2 protein levels in the hippocampus CA1 region after GCI

图6 电镜下观察各组大鼠海马CA1区PUMA和GFAP以及Bcl2的表达(IHS)Figure 6 Representative images of Immunofluorescence staining for PUMA and GFAP, as well as BCL2 in the Hippocampal CA 1 region in the indicated groups (IHS)
3 讨 论
课题组以往的研究发现,雌激素受体调节剂如17β雌二醇、G1均可诱导雌激素膜受体GPR30激活,从而降低脑损伤后神经元凋亡,其分子机制与快速上调AKT、ERK促生存信号通路和抑制神经炎症损伤密切相关[6,16-17]。本研究采用大鼠GCI模型,进一步研究说明GPR30激活的神经保护作用可能与抑制P53-PUMA/Bcl2凋亡信号通路相关。
本研究发现P53特异抑制剂Pifi-α可有效降低I/R 7d海马CA1区神经元凋亡,而GPR30特异拮抗剂G36阻断了Pifi-α的此神经保护作用。
结果提示,转录因子P53可能与GPR30介导的神经保护作用相关。为进一步验证此结论,课题组观察了GPR30激动剂G1对P53磷酸化(激活)和蛋白水平的影响。Western blot及免疫组化结果发现,在缺血再灌注1d和3d,海马CA1区P53磷酸化水平较假手术组控制组显著升高;G1处理降低了GCI诱导的P53磷酸化激活;且p-P53蛋白主要定位在细胞核;P53蛋白水平在各组差异无统计学意义。P53为调控细胞凋亡的重要转录因子,对氧化应激、DNA损伤极为敏感,其稳定和活性在细胞凋亡中至关重要。在GCI模型,P53敲除可降低海马CA1区神经元凋亡[18]。缺血预处理是激发机体内源性保护机制的重要措施;最近研究发现脑缺血预处理可降低再灌注1d和2d海马CA1区神经元细胞核中p-P53水平[19]。Vecino等[20]也发现,在体内、外缺血预处理模型,海马神经元中泛素E3连接酶MDM2蛋白水平升高,从而促进P53泛素化降解和P53的磷酸化激活,促进其细胞核输出,降低神经元损伤。脑外伤后5h给予大鼠Pifi-α可显著降低海马CA1区和DG区P53蛋白水平,降低神经元损伤,改善认知功能[13]。已有研究证明,P53在Ser15位点磷酸化会损伤其与MDM2的结合,从而增加P53的稳定和积累[21]。说明MDM2蛋白也是P53蛋白稳定的重要调控靶点。雌激素受体α激动剂PPT可上调MDM2蛋白水平,改善APP/PS1转基因小鼠认知功能[22]。基于以上文献报道,本研究结果说明,在大鼠GCI模型,GPR30的抗凋亡作用与P53蛋白的稳定与激活密切相相关。
研究证明,P53可通过其转录和/或非转录依赖的信号通路促进细胞凋亡,两条均通过调控线粒体Bcl2家族成员诱导细胞凋亡[23]。P53转录激活可促进其下游靶基因PUMA、Bax、Noxa等促凋亡蛋白表达;而P53非转录依赖通路多为P53转位到细胞浆或线粒体干扰Bax与抗凋亡蛋白Bcl-xL结合,直接诱导线粒体凋亡途径。为进一步揭示P53信号在GPR30神经保护作用机制,课题组首先检测了G1对PUMA、抗凋亡蛋白Bcl2表达的影响。结果显示,G1降低了海马CA1区GCI诱导的PUMA水平,同时增加了Bcl2蛋白水平,并且Bcl2蛋白水平显著高于假手术组对照组;提示G1处理不但起到神经保护作用,而且可激活抗凋亡信号通路;这与我们以往的研究一致,研究发现用G1激活GPR30可上调AKT、ERK促生存信号通路[17]。另外,免疫组化结果显示,PUMA主要定位在细胞核,且在I/R 3d组的海马CA1区可见PUMA与星形胶质细胞标志蛋白GFAP共定位,提示PUMA也可能参与缺血后胶质细胞激活和损伤的调控。进一步研究发现,P53特异抑制剂Pifi-α的影响同G1的作用,均可显著降低GCI后海马CA1区PUMA蛋白水平,同时增加了Bcl2水平。研究结果进一步说明,GPR30激活可能通过抑制P53转录水平上调了抗凋亡蛋白Bcl2表达,同时抑制了促凋亡蛋白PUMA蛋白水平;当然,不能排除G1通过AKT等快速信号通路直接抑制Bcl2依赖的线粒体凋亡。与本研究结果一致,在小鼠抗氧化酶5基因沉默模型中,线粒体解偶联剂鱼藤酮通过增加P53磷酸化激活,上调PUMA了蛋白水平,从而增加胆碱能神经元凋亡[25]。Pifi-α是P53活性抑制剂,可逆性抑制P53依赖的转录活性。在脑卒中、脑外伤动物模型,Pifi-α可抑制P53下游促凋亡靶基因PUMA、BAX等蛋白表达,从而降低神经元损伤[13,27]。
GPR30为雌激素膜受体,课题组以往研究发现,在GCI模型,结合雌激素EDC和E2-BSA可通过GPR30激活介导AKT、ERK1/2快速促生存信号通路[28];另外,用G1激活GPR30也可通过上调IL1RA蛋白表达降低神经炎症损伤[5]。也有文献报道,GPR30在细胞核也有表达,激活抗氧化转录因子Nrf2信号通路[29]。那么,GPR30介导的非基因和/或基因信号通路如何调控P53-PUMA信号,其精确的分子机制尚有待进一步研究。
