大肠杆菌氨基转移酶的非天然氨基酸底物特异性研究
2021-07-12何婷倪雪晨张萍萍陈茹宋晶晶雷佳王行国
何婷,倪雪晨,张萍萍,陈茹,宋晶晶,雷佳,王行国
(湖北大学生命科学学院, 湖北 武汉 430062)
0 引言
氨基转移酶(aminotransferase)也称为转氨酶,催化L-氨基酸和α-酮酸之间的氨基转移[1].反应过程中需要磷酸吡哆醛(PLP)作为辅酶,且在多数情况下使用L-谷氨酸作为氨基供体.氨基转移酶拥有宽泛的底物特异性和较高的转换效率,已被作为生物催化剂广泛用于L-型氨基酸的工业化生产[2]. 氨基转移酶的转氨反应机制为双底物乒乓反应机制,反应是可逆的,其平衡常数为1.0左右[3-4]. 大肠杆菌 (Escherichiacoli) 拥有4种氨基转移酶,分别为天冬氨酸转氨酶(aspartate aminotransferase, AspAT),芳香族氨基酸转氨酶(aromatic amino acid transaminase, TyrAT),支链氨基酸转氨酶(branched-chain amino acid aminotransferase, IlvAT)和丙缬氨酸转氨酶(alanine-valine aminotransferase, AvtAT).它们通常由2或6个亚基组成,分子量为86~182 kD. 单个亚基的分子量为34~46 kD,氨基酸残基数为309~417,且pI值在4.6~5.83之间[5-9].
非天然氨基酸是指天然蛋白质中20种天然氨基酸之外的氨基酸,包括L-非天然氨基酸和D-氨基酸. 由于它们的结构和功能多样性,非天然氨基酸被广泛应用于合成化学的手性骨架和分子支架,合成蛋白酶抑制剂、脑啡呔、人体激素类似物、烟酰胺乙酰胆碱受体和治疗老年痴呆症、高血压、止痛、治肿瘤和抗癫痫的药物[10-14]. 利用非天然氨基酸也能合成新型甜味剂或用于化妆品. 德国赢固赛公司与荷兰DSM公司是少数几个既能生产天然氨基酸又能生产非天然氨基酸的大公司. 目前人工合成出30多种非天然氨基酸,可作为合成活性药物的重要原料.
非天然氨基酸不是氨基转移酶的天然底物. 使用强制进化(forced evolution)的方法能使大肠杆菌细胞利用非天然氨基酸作底物并催化L-谷氨酸和α-酮酸之间的氨基转移[15].这个结果提示大肠杆菌氨基转移酶能催化非天然氨基酸的合成. 本研究的目的是利用大肠杆菌4种氨基转移酶作材料,试图了解氨基转移酶的非天然氨基酸底物特异性. 在此基础上,使用分子生物学方法突变氨基转移酶基因,改进氨基转移酶的活性以提高催化非天然氨基酸的效率.使用易错PCR的方法对酪氨酸转氨酶(TyrAT)基因(tyrB)进行突变,筛选出10突变体.在这10个突变酶中,酶蛋白210位都出现Cys替换Phe. 与野生型酶比较,酪氨酸转氨酶的F210C突变改变酶的催化反应最适温度有提高酶在37 ℃的催化效率.
1 实验材料与方法
1.1 实验菌株、载体和试剂实验中使用的菌株及质粒载体见表1. 细菌通常在LB培养基(胰蛋白胨10 g、NaCl 10 g、酵母提取物5 g,加水至1 L, pH 7.2)中37 ℃培养, -80 ℃甘油冷冻贮藏.质粒载体通常转化至E.coliDH5a或E.coliTop10细菌中扩增或保存.有机和无机试剂均购自上海中科公司. 抗生素、培养基购自Oxoid公司. ExTaq DNA 聚合酶、T4 DNA 连接酶和pMD18-T载体连接试剂盒购自大连宝生物工程有限公司. DNA 凝胶回收试剂盒、PCR 清洁试剂盒购自Axygen 公司.DL-高苯丙氨酸、L-正亮氨酸、L-正缬氨酸、L-叔亮氨酸、L-新戊基甘氨酸、D-正亮氨酸、磷酸氢二钠、磷酸二氢钾、硫酸镁、INT、PES购自Sigma公司;葡萄糖、Tris碱、溴化乙锭、十二烷基硫酸钠(SDS)、琼脂糖、IPTG、氨苄青霉素购自Amresco 公司.
1.2 基因克隆参照分子克隆手册[16]介绍的方法进行基因克隆. 首先从E.coliDH5a细菌中抽提总DNA,然后用总DNA为模板、表2列出的寡聚核苷酸序列作为引物和TaqDNA聚合酶进行PCR反应. PCR反应步骤为: 94 ℃变性5 min后, 再在94 ℃ 变性30 s、53 ℃ 复性90 s、72 ℃ 延伸60 s条件下扩增30个循环, 最后在72 ℃延长5 min. 使用PCR清洁试剂盒回收PCR产物. 将PCR产物直接连接至pMD18-T载体,并转化CaCl2处理的E.coliDH5α感受态细胞,最后在含有氨苄青霉素的LB平板上筛选出抗氨苄青霉素的阳性克隆. 阳性克隆在含有氨苄青霉素的LB培养基中37 ℃培养8 h后抽提质粒DNA. 抽提的质粒DNA经NdeI和XhoI双酶切,鉴定pMD18-T载体是否真正插入外源的DNA片段. 酶切验证后送重组子质粒进行基因测序.
1.3 酶蛋白表达与纯化用NdeI和XhoI双酶切插入目的基因的pMD18-T重组子质粒DNA. 8% 琼脂糖凝胶电泳后,切取目的基因条带, DNA 凝胶回收试剂盒回收DNA片段. 将回收DNA片段与pET23a混合,加入T4 DNA连接酶,16 ℃连接过夜,形成pET23a重组质粒. 将连接后的重组质粒DNA转化CaCl2处理的E.coliBL21(DE3) plysS感受态细胞.来自Paul C Engel实验室的Pagdh基因则插入ptac85形成ptac85重组质粒,转化CaCl2处理的E.coliTop10感受态细胞. 转化后的细菌涂布在含有氨苄青霉素的LB平板上,37 ℃培养过夜,筛选出抗氨苄青霉素的阳性单个菌落. 挑单菌落并接入5 mL的LB液体培养基中37 ℃培养活化.当活化的细菌长到对数期时,按1∶100的比例接入500 mL的LB液体培养基中37 ℃摇床培养.待细菌长到对数期(OD600=0.6~0.8)时加入0.5 mmol/L IPTG,诱导4~8 h.
将培养过夜的细菌4 ℃、6 000 r/min高速冷冻离心15 min,收集的菌体用50 mmol/L Tris-Cl缓冲液(pH 8.0)洗3次,然后重悬于60 mL含1 mol/L DTT和5%甘油的50 mmol/L Tris-Cl缓冲液,并在40 W功率,占空比30%的条件下超声波破碎细胞25 min. 4 ℃、12 000 r/min离心25 min,收集上清液(即粗酶).进行10%SDS-PAGE和考马斯亮兰染色检测目的基因的表达.
使用离子交换层析、疏水层析和凝胶过滤层析三步法纯化酶蛋白. 首先,参照文献[16-18]使用离子交换层析进行第一步纯化. 具体步骤如下: 用含50 mmol/L Tris-Cl (pH 8.0)平衡Q-sepharose FF阴离子交换层析柱,流速控制在2 mL/min. 上样后用0~0.5 mol/L 的NaCl浓度梯度洗脱,自动收集器收集样本,紫外分光光度计检测每管的A280值. 层析完成后收集A280峰值处的蛋白并用10% SDS-PAGE检测目的蛋白. 收集含目的蛋白的样本,分别在TyrAT、IlvAT、AspAT和AvtAT蛋白样品中缓慢地加入40%、30%、45%和35%的硫酸铵,4 ℃保存备用. 第二步纯化则使用疏水层析. 参照文献[17-20], 分别用含40%、30%、45%和35% 硫酸铵的50 mmol/L Tris-HCl(pH 8.0)缓冲液平衡Butyl-Sepharose 4B柱,流速为1 mL/min. 上样后分别用40%~0%、30%~0%、45%~0%和35%~0% 硫酸铵的梯度缓冲液洗脱并自动收集4种氨基转移酶. 依照紫外分光光度计检测的A280洗脱图谱, 10% SDS-PAGE检测目的蛋白并收集目的蛋白样本. 最后用50 mmol/L Tris-HCl(pH 8.0)透析过夜,4 ℃保存备用. 第三步纯化使用凝胶过滤层析[16-17,21], 用含有5 mmol/L NaCl的Tris-Cl(pH 8.0)平衡Sepharose 4 FF层析柱, 流速控制在0.5 mL/min. 上样后继续用含有5 mmol/L NaCl的Tris-Cl洗脱,自动收集和检测A280.用10% SDS-PAGE检测目的蛋白并收集目的蛋白样本, 并加15%甘油后置-80 ℃冷冻冰箱中保藏.
纯化的酶蛋白浓度使用分光光度计进行测定,依照公式C(mg/mL)=(1.55×A280-0.76×A260)×稀释倍数来估算酶蛋白浓度(mg/mL).
1.4 酶活性测定由于氨基酸和酮酸都没有可供检测的光谱,只能用水合茚三酮染色鉴定氨基酸[22].利用光谱检测转氨酶活力则需要偶联另外一个酶反应,如将脱氢酶反应与转氨酶反应偶联起来即可测定NADH在340 nm处的吸收值(如图1所示). 实验中使用的脱氢酶为PaGDH(嗜温性厌氧型链球菌(Peptostreptoccusasaccharolyticus)谷氨酸脱氢酶). 图1还显示,如在酶反应中加入PES(吩嗪乙基硫酸盐)和INT(碘硝基氯化四氮唑蓝),生成紫红色产物甲臜(formazan). 甲臜在512 nm处有最大的吸收值, 因此也可以用96孔板检测转氨酶活性.

图1 脱氢酶与转氨酶偶联的酶催化反应示意图
依据上述原理,可用3种方法进行酶活性定性或定量测定. 一种是分光光度计法,即在1 mL反应体系中, 3 mmol/L非天然氨基酸、2 mmol/Lα-酮戊二酸、2 μmol/L PLP、1 mmol/L NAD+、5 μL 0.2 mg/mL PaGDH、50 mmol/L Tris-HCl(pH 8.0)混匀后加入转氨酶,并在Shmadzu UV-2550分光光度计上37 ℃、340 nm处测定酶反应初速度[23]. 另一种是酶标仪检测法,即在上述1 mL反应体系中再加0.04 mg/mL INT和0.3 mg/mL PES. 96孔板每个孔中加入200 μL 反应液,混匀后37 ℃培养箱中放置10 min, BioTek酶标仪测定512 nm吸光值或直接拍照. 最后一种方法是TLC法[15],即用正丁醇∶冰乙酸∶dH2O (4∶1∶3) 预处理Silica gel 60硅胶板. 每个样品取3 μL反应混合物点样到硅胶板上,然后将硅胶板置于盛有正丙醇∶dH2O(7∶3,V/V) 展层液的层析缸中进行TLC.TLC完成后取出硅胶板并在45 ℃烘干,最后用0.5%水合茚三酮乙醇溶液喷雾显色.氨基酸与茚三酮反应生成紫红色斑点,因此TLC法可用来进行氨基转移酶活性的定性检测.
1.5 易错PCR与定向进化筛选以实验室构建好的pET23a-tyrB质粒DNA为模板,使用引物tyrB-F和tyrB-R进行易错PCR (sequential error-prone PCR) 扩增. 通过调整PCR反应条件,如提高镁离子浓度、添加锰离子、改变体系中4种dNTPs浓度和运用低保真度DNA聚合酶等来改变扩增过程中的突变频率,从而以一定的频率向目的基因中随机引入突变,获得蛋白质分子的随机突变体.参照文献[24-25]的方法,进行易错PCR. PCR反应程序为:94 ℃变性5 min,94 ℃ 30 s、59 ℃ 1.5 min、72 ℃ 1 min扩增30个循环并72 ℃延伸7 min. PCR反应产物用0.7 %琼脂糖凝胶电泳分析.回收的PCR产物溶于40 μL无菌超纯水并在-20 ℃保存备用.
将改变镁离子、锰离子和4种dNTPs浓度的PCR产物回收后混合,用NdeI和XhoI双酶切, 插入pET23a质粒并转化E.coliBL21(DE3) plysS,建立突变文库(5 000菌落). 将突变文库的细菌接种到以5 mmol/LDL-高苯丙氨酸或5 mmol/LD-正亮氨酸为氮源的M9培养基中培养24 h. 再按1∶100接种至含有3 mmol/LDL-高苯丙氨酸或3 mmol/LD-正亮氨酸为氮源的M9培养基中再培养24 h. 然后按1∶100接种至含有1 mmol/LDL-高苯丙氨酸或1 mmol/LD-正亮氨酸为氮源的M9培养基中培养48 h.通过3轮定向进化筛选后, 涂1 mmol/LDL-高苯丙氨酸或1 mmol/LD-正亮氨酸为氮源的M9培养基平板,收集筛选出的单个菌落,抽提重组子质粒,靶基因DNA测序确定突变位点,并进行酶蛋白表达和纯化以及酶活性分析.
2 结果与分析
2.1 4种氨基转移酶基因克隆、酶蛋白表达纯化与酶活性经PCR反应后,成功获得大肠杆菌4种氨基转移酶基因aspC、ilvE、avtA和tyrB.其基因大小分别为1 191 bp、930 bp、1 254 bp和1 194 bp. 将它们分别克隆至pET23a表达质粒并转化E.coliBL21(DE3) plys S, 表达的酶蛋白大小分别为43 kD、33.5 kD、46.7 kD和43 kD,与基因库报道的大小一致. 4种氨基转移酶,即天冬氨酸氨基转移酶(AspAT)、丙缬氨酸氨基转移酶(AvtAT)、支链氨酸氨基转移酶(IlvAT)和酪氨酸氨基转移酶(TyrAT),经三步层析分离纯化,每L细菌培养物可得到30~58 mg纯蛋白. 10% SDS-PAGE显示纯化后的4种酶蛋白纯度达到电泳纯 (见图2).
依据氨基转移酶催化氨基酸和α-酮戊二酸生成α-酮酸和谷氨酸的酶反应机理,设计酶反应体系检测纯化的4种氨基转移酶的活性. 在酶反应体系中,分别用天冬氨酸、亮氨酸、丙氨酸/缬氨酸、酪氨酸和α-酮戊二酸作AspAT、IlvAT、AvtAT、TyrAT的底物, PLP(磷酸吡哆醛)为辅酶.通过TLC检测产物谷氨酸的生成,判断纯化的氨基转移酶是否具有酶活性.TLC的结果显示,4种氨基转移酶都具有转氨活性(见图3).

1:AspAT; 2:IlvAT; 3:AvtAT; 4:TyrAT图2 4 种大肠杆菌纯酶蛋白的10% SDS-PAGE

1和2:氨基酸标准品; 3:未加酶的负对照; 4:酶反应物.(a) AspAT; (b) IlvAT; (c) AvtAT; (d) TyrAT图3 4 种大肠杆菌氨基转移酶的酶活性TLC分析
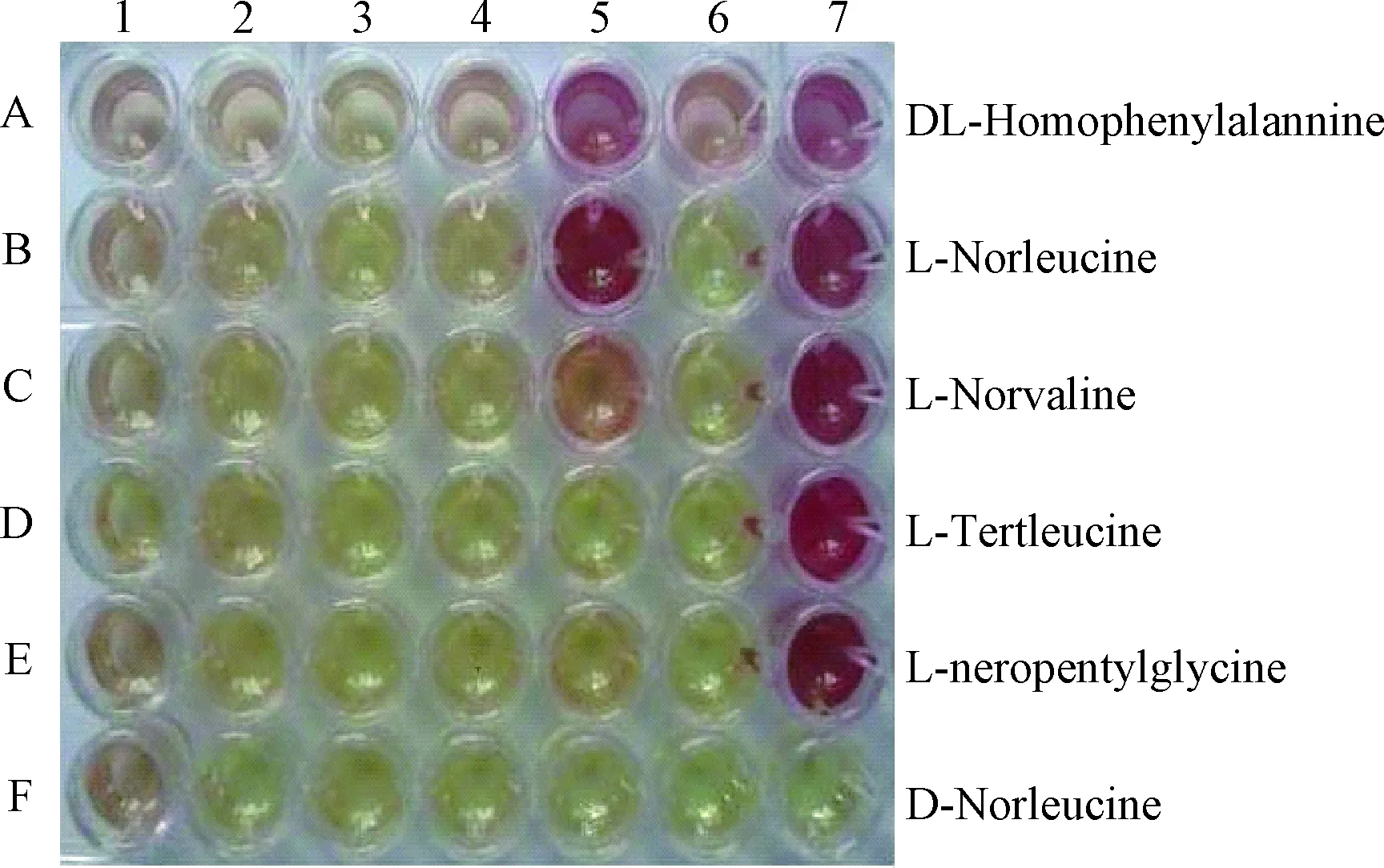
1:空白对照;2和3分别为E. coli BL21(DE3)粗酶液和含pET23a质粒的E. coli BL21(DE3)粗酶液; 4. AvtAT纯酶;5.TyrAT纯酶; 6. AspAT纯酶; 7. IlvAT纯酶.图4 4种氨基转移酶对6种非天然氨基酸底物特异性
2.2 4种氨基转移酶对6种非天然氨基酸的催化活性使用氨基转移酶偶联谷氨酸脱氢酶反应的方法,通过甲臜的颜色反应调查4种氨基转移酶催化6种非天然氨基酸(DL-高苯丙氨酸、L-正缬氨酸、L-正亮氨酸、D-正亮氨酸、L-叔亮氨酸和L-新戊基甘氨酸)的酶活性,判断它们的非天然氨基酸底物特异性. 图4的结果显示: 在37 ℃、pH 8.0条件下,AvtAT基本不催化6种非天然氨基酸的氨基转移,而AspAT仅对DL-高苯丙氨酸显示极微弱的催化反应. TyrAT能用DL-高苯丙氨酸、L-正亮氨酸和L-正缬氨酸作底物进行的氨基转移反应, 而IlvAT则催化DL-高苯丙氨酸、L-正缬氨酸、L-正亮氨酸、L-叔亮氨酸和L-新戊基甘氨酸的氨基转移. 图4的甲臜颜色反应结果与TLC法测得的结果(未显示)一致.
2.3 酪氨酸氨基转移酶基因突变与突变酶的非天然氨基酸底物特异性4种氨基转移酶对6种非天然氨基酸的催化活性检测发现,TyrAT仅催化DL-高苯丙氨酸、L-正亮氨酸和L-正缬氨酸的氨基转移. 因此,我们选择TyrAT进行基因突变,试图扩大这个酶的非天然氨基酸底物特异性.易错PCR后获得5 000个单菌落的突变体文库,然后使用DL-高苯丙氨酸和L-正亮氨酸作碳源进行定向进化筛选. 从筛选出的菌落中抽提重组质粒DNA并进行靶基因测序,获得的结果见图5. 图5显示: 使用DL-高苯丙氨酸作氮源定向进化,筛选出6种不同的氨基酸变异突变体,其中H-14突变体催化DL-高苯丙氨酸氨基转移的活性最高. 在用L-正亮氨酸作氮源定向进化筛选中仅获得4种不同的氨基酸变异突变体,其中N-19突变体催化L-正亮氨酸氨基转移的活性最高. 粗酶液活性分析发现H-14突变体的DL-高苯丙氨酸催化活力比野生型提高5倍,而N-19突变体的L-正亮氨酸催化活力比野生型提高14.6 倍. 在两种非天然氨基酸的定向进化筛选中,所有的突变体都在F210位上出现了半胱氨酸(Cys)的替换. TLC和96孔板颜色反应检测10个突变酶对6种非天然氨基酸的催化反应,发现它们仍与野生型酶一样,仅催化DL-高苯丙氨酸、L-正亮氨酸和L-正缬氨酸的氨基转移,并未改变该酶的非天然氨基酸底物特异性.

图5 易错PCR突变体文库中筛选的TyrAT突变体
2.4 F210C突变酶的酶学特性鉴于所有的突变体都在Phe210位上出现了半胱氨酸(Cys)的替换,我们对F210C突变的作用进行了深入的探讨. H-40突变体酪氨酸氨基转移酶基因上有3位点的碱基改变,135位的A突变为G、629位的T突变为G以及843位的C突变为T. A135G和C843T为同义突变,酶蛋白相应位点Q45和R281的氨基酸仍未改换. 只有T629G位点突变导致酶蛋白相应位点210上的Phe被替换成Cys. 因此,我们选用H-40突变酶来研究Phe210Cys的酶学特性,探讨210位Cys替换Phe对酶活性的影响.
在不同的pH (7.0、7.5、8.0、8.5) 和不同的温度(25、30、37、45、50、55 ℃) 条件下,使用分光光度计法检测野生型和F210C突变酶催化DL-高苯丙氨酸、L-正亮氨酸和L-正缬氨酸的转氨反应.结果发现野生型和F210C突变酶在不同pH条件下没有显著性差异,而温度变化则影响两种酶催化3种非天然氨基酸的转氨反应. 图6显示了野生型和F210C突变酶催化DL-高苯丙氨酸、L-正亮氨酸和L-正缬氨酸转氨反应的结果. 野生型TyrAT催化DL-高苯丙氨酸和L-正缬氨酸转氨反应的最适温度为45 ℃,催化L-正亮氨酸的最适温度为37 ℃. 与野生型酶不同,F210C突变酶催化DL-高苯丙氨酸和L-正缬氨酸转氨反应的最适温度为37 ℃,仅催化L-正亮氨酸的最适温度仍为37 ℃. 此外, F210C突变酶在37 ℃催化DL-高苯丙氨酸和L-正缬氨酸的转氨活性明显比野生型高(见图6中(a)和(c)). 分光光度计法检测的结果显示,F210C突变酶37 ℃催化DL-高苯丙氨酸转氨的酶活力为1.36 U/mg, 是野生型酶的酶活力(0.79 U/mg)的1.72倍. 这些结果表明TyrAT 210位Phe突变为Cys显著地改变了酶的催化反应最适温度.

图6 温度对野生型和F210C突变酶催化DL-高苯丙氨酸、L-正亮氨酸和L-叔亮氨酸转氨反应速率的影响
3 讨论
本实验使用PCR技术,成功从大肠杆菌基因组DNA中克隆出4种氨基转移酶基因aspC、ilvE、avtA和tyrB.克隆的基因插入pET23a表达质粒、转化E.coliBL(DE3) plysS和IPTG诱导表达后,使用离子交换层析、疏水层析和凝胶过滤层析三步法纯化,获得30~58 mg/L电泳纯酶蛋白. 4种氨基转移酶(AspAT、AvtAT、IlvAT、TyrAT)对6种非天然氨基酸底物特异性检测发现,4种氨基转移酶中只有IlvAT催化DL-高苯丙氨酸、L-正缬氨酸、L-正亮氨酸、L-叔亮氨酸和L-新戊基甘氨酸的氨基转移,而TyrAT仅催化DL-高苯丙氨酸、L-正亮氨酸和L-正缬氨酸的氨基转移.其他2种氨基转移酶对6种非天然氨基酸无催化活性.
使用易错PCR突变tyrB基因并建立突变体文库后,用DL-高苯丙氨酸和L-正亮氨酸作氮源进行定向进化筛选,共获得10个不同位点突变的TyrAT突变酶. 与野生型酶一样,这些突变酶仅催化DL-高苯丙氨酸、L-正亮氨酸和L-正缬氨酸的氨基转移. 虽然突变酶的酶活性不同于野生型酶但并未改变TyrAT的非天然氨基酸底物特异性. F210C突变酶的酶学特性研究发现,TyrAT中210位的Phe突变为Cys后显著地改变了酶的催化反应最适温度. 野生型TyrAT催化DL-高苯丙氨酸和L-叔亮氨酸的氨基转移反应最适温底为45 ℃,而F210C突变酶催化反应的最适温度为37 ℃. TyrAT 酶210位氨基酸的改变(Phe→Cys)能有效降低催化反应的最适温度. 很明显,利用易错PCR和定向进化技术扩大TyrAT酶的非天然氨基酸底物特异性仍是一个需要深入探索的课题.
图7a显示芳香族氨基酸转氨酶的三维结构,Ser293为该酶的催化基团[26]. 依据野生型芳香族氨基酸转氨酶的晶体结构(PDB ID: 3TAT),利用Discovery Studio 2.5软件进行分析发现,H-14突变酶的L108V和V145M分别位于底物结合位点Ser109和Arg292、Ser296、Ser297附近,而它的N243S突变位点位于辅酶结合位点Ser255和Lys258附近. 这些突变可能有利于DL-高苯丙氨酸在活性袋内结合和辅酶的稳定,从而使突变酶的活力高于野生型.N-19突变酶的T186A突变位点位于底物识别位点Arg386和Phe360附近,氨基酸残基的亲水性和侧链变小可能有利于α-酮酸底物的结合,使突变酶对L-正亮氨酸的催化活力高于野生型. 所有10个突变酶的F210位都发生Cys替换(见图7b),但都没有在S293催化位点发生突变. F210C突变位点位于底物识别位点Asp222和Leu39附近,Cys替换Phe后可能改变活性袋的柔性. 210位的Cys替换Phe有利于非天然氨基酸底物在活性袋内运动,使底物分子容易调整至正确的催化位置,从而降底酶的催化反应最适温度.

图7 芳香族氨基酸转氨酶的三维结构与F210C突变示意图
