HPLC-ELSD法同时测定葡萄糖原料药中6种糖类有关物质的含量
2021-07-11宫兴华郑磊李娟
宫兴华 郑磊 李娟
中图分类号 R917 文献标志码 A 文章编号 1001-0408(2021)08-1241-05
DOI 10.6039/j.issn.1001-0408.2021.10.14
摘 要 目的:建立同时测定葡萄糖原料药中果糖、麦芽糖、异麦芽糖、麦芽三糖、麦芽四糖、麦芽五糖等6种糖类有关物质的方法。方法:采用高效液相色谱-蒸发光散射检测法。色谱柱为XBridge Amide,流动相为乙腈-水(75 ∶ 25,V/V),流速为0.5 mL/min,柱温为30 ℃,进样量为10 μL;检测器为蒸发光散射检测器,载气为氮气,气体压力为40 psi,蒸发温度为80 ℃,漂移管温度为80 ℃,增益为100。结果:上述6种糖类有关物质检测质量浓度的线性范围分别为5.99~59.88、9.90~98.96、9.92~99.19、5.97~59.74、4.03~40.32、5.89~58.89 μg/mL(r>0.999 0);定量限分别为1.5、1.5、1.5、3.0、3.0、3.0 μg/mL,检测限分别为0.5、0.5、0.5、1.0、1.0、1.0 μg/mL;精密度、稳定性(12 h)、重复性试验的RSD均小于2.0%;平均加样回收率分别为95.87%~98.59%(RSD=1.04%,n=9)、95.66%~99.84%(RSD=1.20%,n=9)、96.11%~98.97%(RSD=1.04%,n=9)、95.06%~99.11%(RSD=1.25%,n=9)、95.69%~98.22%(RSD=0.83%,n=9)、95.34%~98.56%(RSD=1.01%,n=9)。9批葡萄糖原料药中上述6种糖类有关物质含量分别为1.26~2.22 、2.55~3.36、2.37~3.37、1.28~2.01、0~2.11、0~1.89 mg/g。结论:所建方法准确性好、灵敏度高,可用于葡萄糖中糖類有关物质的检测。
关键词 葡萄糖;糖类;有关物质;高效液相色谱-蒸发光散射检测法
Simultaneous Determination of 6 Carbohydrate Related Substances in Glucose by HPLC-ELSD
GONG Xinghua1,ZHENG Lei1,LI Juan2(1. Dept. of Pharmacy, Shandong Provincial Third Hospital, Jinan 250031, China; 2. Dept. of Pharmacy, the Second Hospital of Shandong University, Jinan 250033, China)
ABSTRACT OBJECTIVE: To establish the method for the simultaneous determination of 6 carbohydrate related substances in glucose as fructose, maltose, isomaltose, maltotriose, maltotetraose and maltopentaose. METHODS: HPLC-ELSD was adopted. The determine was performed on XBridge Amide column with mobile phase consisted of acetonitrile-water (75 ∶ 25, V/V) at a flow rate of 0.5 mL/min. The column temperature was set at 30 ℃, and the sample size was 10 L. The detector was evaporative light scattering detector, the carrier gas was nitrogen, the gas pressure was 40 psi, the evaporation temperature was 80 ℃, the drift tube temperature was 80 ℃, and the gain was 100. RESULTS: The linear range of 6 carbohydrate related substances were 5.99-59.88, 9.90-98.96, 9.92-99.19, 5.97-59.74, 4.03-40.32, 5.89-58.89 μg/mL(r>0.999 0). The quantitation limits were 1.5, 1.5, 1.5, 3.0, 3.0 and 3.0 μg/mL, respectively. The detection limits were 0.5, 0.5, 0.5, 1.0, 1.0, 1.0 μg/mL, respectively. RSDs of precision, stability (12 h) and reproducibility tests were all lower than 2.0%. The average recoveries were 95.87%-98.59%(RSD=1.04%,n=9), 95.66%-99.84%(RSD=1.20%,n=9), 96.11%-98.97%(RSD=1.04%,n=9), 95.06%-99.11%(RSD=1.25%,n=9),95.69%-98.22%(RSD=0.83%,n=9), 95.34%-98.56%(RSD=1.01%,n=9). The contents of 6 carbohydrate related substances in 9 batches of glucose were 1.26-2.22, 2.55-3.36, 2.37-3.37, 1.28-2.01, 0-2.11 and 0-1.89 mg/g, respectively. CONCLUSIONS: Established method is accurate and sensitive, and can be used for the detection of carbohydrate related substances in glucose.
KEYWORDS Glucose; Carbohydrate; Related substance; HPLC-ELSD
葡萄糖是临床常用的营养药物,且葡萄糖注射液作为静脉用药的溶媒,在临床应用非常广泛。葡萄糖是机体所需能量的主要来源,在机体内可被氧化成二氧化碳和水,同时可为机体提供所需的能量[1],其质量标准收载于2020年版《中国药典》(二部)[2]、9.0版《欧洲药典》[3]、40版《美国药典》[4]和17版《日本药局方》[5]中。葡萄糖作为单糖除可在碱性条件下发生差向异构化而转化为果糖外,还可发生脱水缩合反应转化为二糖和多糖[6]。虽然二糖和多糖成分没有毒性,并不影响葡萄糖使用的安全性,但可影响其纯度,并可在一定程度上反映葡萄糖原料药的品质优劣和制备工艺的先进与否。目前,仅有9.0版《欧洲药典》同时对葡萄糖原料药中果糖、麦芽糖、异麦芽糖和麦芽三糖等4种糖类有关物质进行了规定[2],而其他各国药典均未有此类有关物质的控制项。此外,葡萄糖在发生脱水缩合过程中还有可能生成麦芽四糖和麦芽五糖,而麦芽四糖和麦芽五糖会降低葡萄糖的纯度,从而影响葡萄糖的质量,且在各国药典中也均未见相关检测要求。
由于糖类化合物分子结构中缺少紫外吸收基团,不具有紫外吸收的特征,因此不能采用紫外检测器进行检测。检测糖类化合物时,通常会选择蒸发光散射检测器(ELSD)或者示差折光检测器,而示差折光检测器受试验环境影响较大,且存在稳定性差、灵敏度低的缺点[7]。9.0版《欧洲药典》以钙型强阳离子交换柱为色谱柱,水为流动相,采用示差折光检测器对果糖、麦芽糖、异麦芽糖和麦芽三糖进行检测[3]。本课题组前期研究发现,在上述色谱条件下,麦芽糖和异麦芽糖的相对保留时间均为0.8,不能有效分离。基于此,本研究采用酰胺基色谱柱,以高效液相色谱-蒸发光散射检测法(HPLC-ELSD)同时测定葡萄糖原料药中果糖、麦芽糖、异麦芽糖、麦芽三糖、麦芽四糖、麦芽五糖等6种糖类有关物质,旨在为葡萄糖原料药的质量控制提供参考。
1 材料
1.1 主要仪器
本研究所用主要仪器有ELSD6000 1290型HPLC仪及配套的在线真空脱气装置、四元梯度泵、自动进样器、柱温箱以及ELSD(美国Alltech公司),Mettler XP205型十万分之一电子天平(瑞士Mettler Toledo公司),GZX 9030MBE型电热恒温鼓风干燥箱(上海博讯仪器公司),YB-Ⅱ型澄明度检测仪(天津综科科技公司)等。
1.2 药品与试剂
葡萄糖对照品(批号G173070,纯度99.9%)、果糖对照品(批号G173081,纯度99.6%)、麦芽糖对照品(批号G173121,纯度98.4%)、异麦芽糖对照品(批号G173122,纯度99%)、麦芽三糖对照品(批号G183037,纯度99%)、麦芽四糖对照品(批号G173069,纯度99%)、麦芽五糖对照品(批号G173066,纯度98%)均购自德国Dr. Ehrenstorfer GmbH公司;葡萄糖原料药(中国XR公司,批号19061211、19071811、19101011,纯度大于98.5%;中国LX公司,批号200217、200506,纯度大于98.5%;中国CX公司,批号A0200508、A0200102,纯度大于99.0%;中国LK公司,批号511911081、511912051,纯度大于99.0%);乙腈为色谱纯,其余试剂均为分析纯,水为超纯水。
2 方法與结果
2.1 色谱条件
以XBridge Amide(150 mm×4.6 mm,3.5 μm)为色谱柱,乙腈-水(75 ∶ 25,V/V)为流动相;流速为0.5 mL/min;柱温为30 ℃;进样量为10 μL;检测器为ELSD;载气为氮气;气体压力为40 psi;蒸发温度为80 ℃;漂移管温度为80 ℃;增益为100。
2.2 溶液的配制
2.2.1 葡萄糖对照品贮备液 精密称取葡萄糖对照品适量,置于10 mL量瓶中,用流动相溶解并定容,摇匀,即得质量浓度为100 mg/mL的葡萄糖对照品贮备液。
2.2.2 有关物质对照品贮备液 精密称取果糖、麦芽糖、异麦芽糖、麦芽三糖、麦芽四糖、麦芽五糖对照品各适量,分别置于100 mL量瓶中,用流动相溶解并定容,摇匀,制得质量浓度分别为0.598 8、0.989 6、0.991 9、0.597 4、0.403 2、0.588 9 mg/mL的各有关物质单一对照品贮备液。
2.2.3 供试品溶液 精密称取葡萄糖原料药0.5 g,置于50 mL量瓶中,用流动相溶解并定容,摇匀,即得质量浓度约为10 mg/mL的供试品溶液。
2.2.4 系统适用性试验溶液 精密量取“2.2.2”项下各有关物质单一对照品贮备液1 mL,置于同一20 mL量瓶中,精密加入“2.2.1”项下葡萄糖对照品贮备液2 mL,用流动相稀释至刻度,摇匀,即得系统适用性试验溶液。
2.3 系统适用性试验
取“2.2”项下系统适用性试验溶液、供试品溶液、空白对照溶液(流动相)适量,按“2.1”项下色谱条件进样测定,记录色谱图。结果,各有关物质色谱峰之间以及与主峰之间的分离度均大于1.5(麦芽糖与异麦芽糖色谱峰之间的分离度大于1.5),理论板数以葡萄糖计均大于5 000,且空白对照溶液色谱图中未检测到色谱峰,表明其对所有待测成分无干扰,详见图1。
2.4 破坏试验
2.4.1 溶液配制 (1)酸破坏溶液——取葡萄糖原料药(批号19061211)约0.5 g,置于50 mL量瓶中,精密加入0.1 mol/L盐酸溶液1 mL,室温下放置30 min;精密加入0.1 mol/L氢氧化钠溶液1 mL中和,用流动相稀释至刻度,摇匀,即得酸破坏溶液。(2)碱破坏溶液——取葡萄糖原料药(批号19061211)约0.5 g,置于50 mL量瓶中,精密加入0.1 mol/L氢氧化钠溶液1 mL,室温下放置20 min;精密加入0.1 mol/L盐酸溶液1 mL中和,用流动相稀释至刻度,摇匀,即得碱破坏溶液。(3)氧化破坏溶液——取葡萄糖原料药(批号19061211)约0.5 g,置于50 mL量瓶中,精密加入3%双氧水5 mL,室温下放置2 h,用流动相稀释至刻度,摇匀,即得氧化破坏溶液。(4)高温破坏溶液——取葡萄糖原料药(批号19061211)约0.5 g,置于50 mL量瓶中,于100 ℃电热恒温鼓风干燥箱中加热2 h,冷却后用流动相稀释至刻度,摇匀,即得高温破坏溶液。(5)光照破坏溶液——取葡萄糖原料药(批号19061211)约0.5 g,置于50 mL量瓶中,于4 000 lx光照条件下放置12 h,用流动相稀释至刻度,摇匀,即得光照破坏溶液。
2.4.2 破坏试验 取上述各溶液适量,按“2.1”项下色谱条件进样测定,记录色谱图。结果,葡萄糖原料药在光照条件下较为稳定;在酸、碱、氧化和高温条件下均不稳定,可见较多未知杂质,且在碱性条件下果糖色谱峰响应增强,表明在碱性条件下,有部分葡萄糖可能转化为果糖;葡萄糖原料药经酸、碱、氧化、高温和光照破坏试验后,各有关物质峰之间以及与新产生的杂质峰之间均能完全分离,详见图2。
2.5 线性关系考察
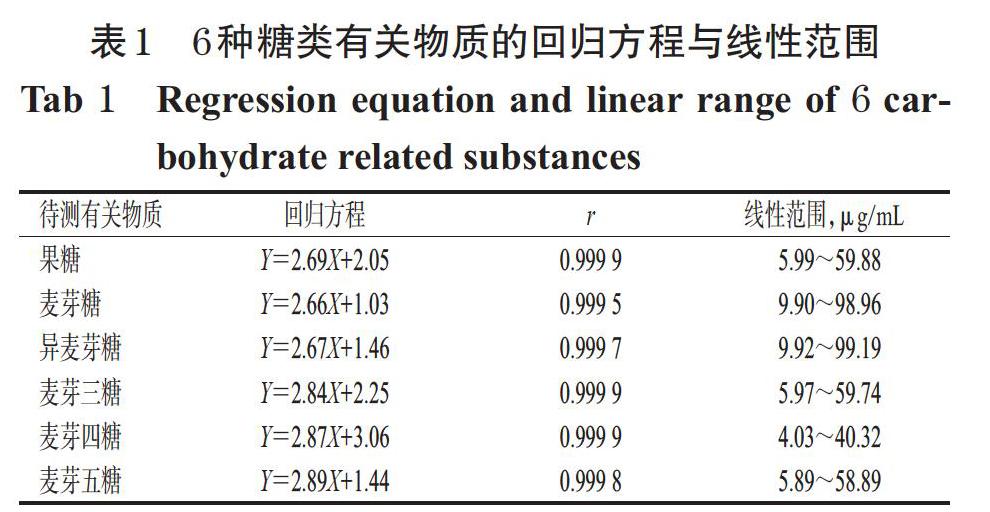
精密量取“2.2.2”项下各有关物质单一对照品贮备液5 mL,置于同一50 mL量瓶中,用流动相稀释至刻度,摇匀,再分别精密量取1、2、4、6、8、10 mL,置于10 mL量瓶中,用流动相稀释至刻度,摇匀,制得果糖质量浓度分别为5.99、11.98、23.95、35.93、47.90、59.88 μg/mL,麦芽糖分别为9.90、19.79、39.58、59.38、79.17、98.96 μg/mL,异麦芽糖分别为9.92、19.84、39.68、59.51、79.35、99.19 μg/mL,麦芽三糖分别为5.97、11.95、23.90、35.84、47.79、59.74 μg/mL,麦芽四糖分别为4.03、8.06、16.13、24.19、32.26、40.32 μg/mL,麦芽五糖分别为5.89、11.78、23.56、35.33、47.11、58.89 μg/mL的系列标准溶液。按“2.1”项下色谱条件进样测定,记录色谱图。以各有关物质质量浓度的对数值(X)为横坐标、峰面积的对数值(Y)为纵坐标进行线性回归,结果见表1。
2.6 定量限与检测限考察
取“2.5”项下最低质量浓度的系列标准溶液,用流动相逐步稀释,按“2.1”项下色谱条件进样测定,以信噪比10 ∶ 1、3 ∶ 1分别计算定量限与检测限。结果,果糖、麦芽糖、异麦芽糖、麦芽三糖、麦芽四糖、麦芽五糖的定量限分别为1.5、1.5、1.5、3.0、3.0、3.0 μg/mL,检测限分别为0.5、0.5、0.5、1.0、1.0、1.0 μg/mL。
2.7 精密度试验
取“2.2.4”项下系统适用性试验溶液,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,果糖、麦芽糖、异麦芽糖、麦芽三糖、麦芽四糖、麦芽五糖峰面积的RSD均小于2.0%(n=6),表明仪器精密度良好。
2.8 稳定性试验
取“2.2.3”项下供试品溶液(批号19061211),分别于室温下放置0、2、4、8、12 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果,果糖、麦芽糖、异麦芽糖、麦芽三糖、麦芽四糖、麦芽五糖峰面积的RSD均小于2.0%(n=5),表明供试品溶液于室温下放置12 h内稳定性良好。
2.9 重复性试验
取葡萄糖原料药(批号19061211)约0.5 g,共6份,按“2.2.3”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并按标准曲线法计算样品中各有关物质的含量。结果,果糖、麦芽糖、异麦芽糖、麦芽三糖、麦芽四糖、麦芽五糖含量的RSD均小于2.0%(n=6),表明方法重复性良好。
2.10 加样回收率试验
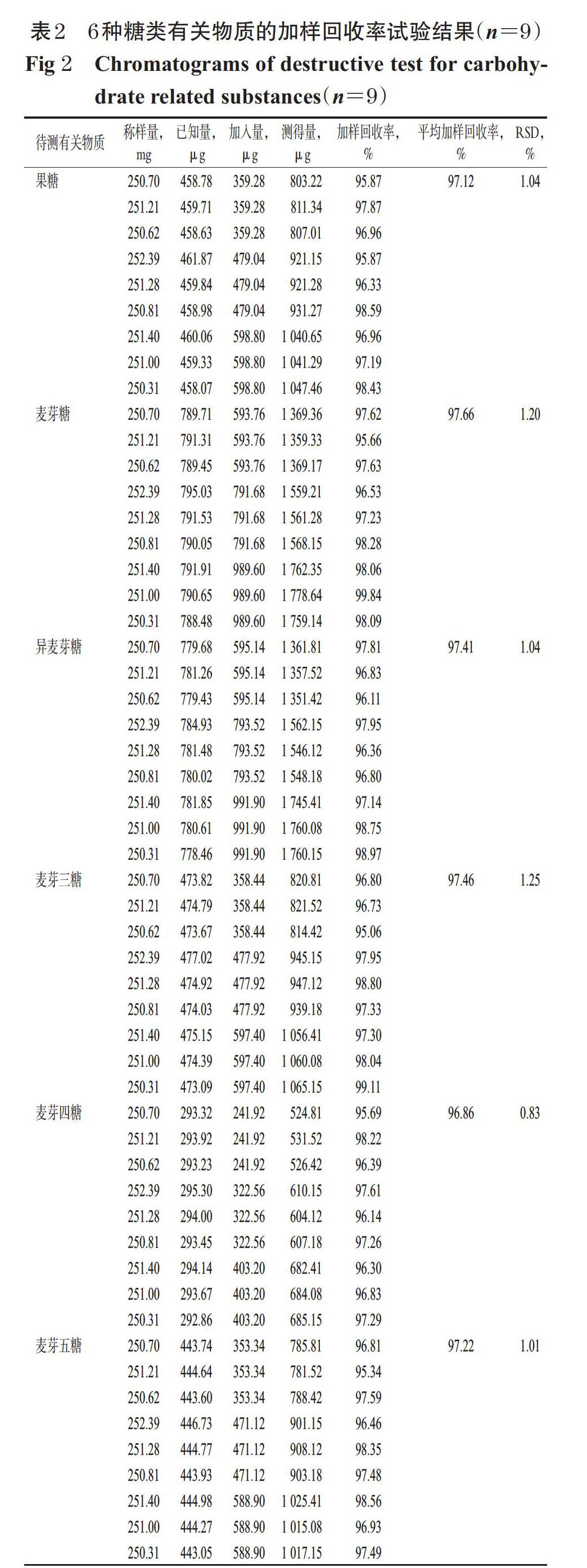
精密称取葡萄糖原料药(批号19061211)约250 mg,共9份,分别置于50 mL量瓶中,精密加入“2.2.2”项下各有关物质单一对照品贮备液600、800、1 000 μL,按“2.2.3”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算加样加收率,结果见表2。
2.11 耐用性试验
取“2.2.4”项下系统适用性试验溶液,按“2.1”项下色谱条件以不同色谱柱[XBridge Amide(150 mm×4.6 mm,3.5 μm)、Inert Sustain Amide(100 mm×4.6 mm,3 μm)、Inertsil Amide(150 mm×4.6 mm,3.5 μm)]、不同柱溫(25、30、35 ℃)、不同流速(0.4、0.5、0.6 mL/min)进行分析。结果,当以上条件发生变化时,各色谱峰均符合耐用性试验的要求,测得各成分含量的RSD均小于2.0%(n=3)。
2.12 样品中6种糖类有关物质的测定
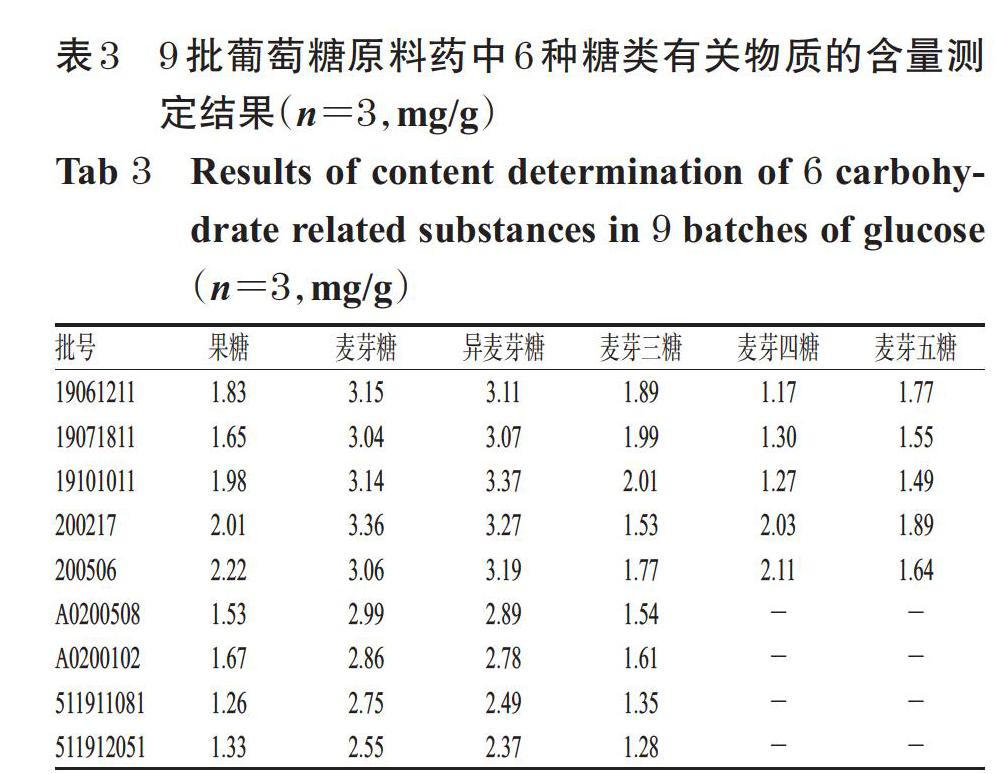
取9批葡萄糖原料药适量,按“2.2.3”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并按标准曲线法计算样品中6种有关物质的含量。平行操作3次,取平均值,结果见表3(表中,“-”表示未检出)。
3 讨论
9.0版《欧洲药典》采用钙型强阳离子交换色谱柱进行检测,但该色谱柱并不能将麦芽糖和异麦芽糖分开[3]。因此,本课题组拟选择正相色谱柱进行分析。首先,对氨基色谱柱进行考察,发现因氨基色谱柱所需平衡时间较长,使得各色谱峰存在保留时间漂移的现象。有研究认为,酰胺柱以高纯硅胶为基质键合烷基酰胺基,是对极性化合物具有强保留能力的亲水性色谱柱[8-10]。因此,本课题组对酰胺柱进行考察,发现该色谱柱可避免氨基色谱柱保留时间漂移和基线不稳定的缺点,且待测物质尤其是麦芽糖和异麦芽糖可以实现基线分离。
本课题组前期分别比较了不同流动相系统[甲醇-水(80 ∶ 20,V/V)、甲醇-0.1%甲酸水溶液(80 ∶ 20,V/V)、乙腈-水(75 ∶ 25,V/V)、乙腈-0.1%甲酸水溶液(75 ∶ 25, V/V)]的分离效果。结果,以0.1%甲酸水溶液作为水相时,各待测成分色谱峰存在不同程度的拖尾现象,峰形较差;以乙腈-水为流动相时,各色谱峰分离度更好且优于甲醇-水,因此本研究选择乙腈-水为流动相。
本研究对示差折光检测器和ELSD两种检测器进行了考察,虽然上述两种检测器均为通用型检测器,均可以对糖类化合物进行检测,但是示差折光检测器在使用前需要较长的平衡时间,且易受环境的干扰,温度、压力、流速的变化均会引起待测物质密度变化,进而导致折射率的改变,检测灵敏度较ELSD低[11-13]。因此,本研究选用ELSD进行检测,加之本研究采用乙腈-水作为流动相系统,不含有非挥发性盐类成分,正适用于ELSD[12]。
ELSD由雾化器、加热漂移管和光散射池组成,流动相在雾化器入口处被氮气雾化形成气溶胶,进入加热漂移管中,其中的溶剂被蒸发掉,剩余的样品溶质进入检测池[14-15]。而载气压力和漂移管温度对检测结果的影响较大[16-17]。因此,本课题组对载气(氮气)压力和漂移管温度进行了考察。结果,当载气压力较低时,流动相雾化不充分;当载气压力较高时,导致待测成分损失,色谱响应减弱,故分别于不同载气压力(30、35、40、45、50 psi)下检测各色谱峰的响应强度和基线噪声[16]。结果,当载气压力为40 psi时,各待测成分色谱峰的信噪比最高。漂移管温度对色谱峰的影响主要体现在基线噪声上,当漂移管温度较低时,溶剂挥发不完全,基线不稳;当漂移管温度较高时,基线噪声增加[17]。因此,本课题组又分别对不同漂移管温度(70、75、80、85、90 ℃)进行了考察。结果,当漂移管温度为80 ℃时,基线平稳,噪声信号较弱。
2020年版《中国药典》(二部)检测标准并未规定葡萄糖中的糖类有关物质的检测方法及限度,存在检测项目不完善的不足,使得国内的葡萄糖原料药生产企业暂未关注该类有关物质[1]。本研究中,9批葡萄糖原料药中6种糖类有关物质含量分别为果糖1.26~2.22 mg/g、麦芽糖2.55~3.36 mg/g、异麦芽糖2.37~3.37 mg/g、麦芽三糖1.28~2.01 mg/g、麦芽四糖0~2.11 mg/g、麦芽五糖0~1.89 mg/g。按照9.0版《欧洲药典》中的限度要求,即果糖不得过0.15%、麦芽糖与异麦芽糖总量不得过0.4%、麦芽三糖不得过0.2%[3],本研究中有部分批次样品中的糖类有关物质超出了上述标准。
综上所述,本研究首次建立了同时测定葡萄糖原料药中6种糖类有关物质的HPLC-ELSD法,该方法解决了9.0版《欧洲药典》中麦芽糖和异麦芽糖不能分离的缺点;所建方法准确性好、灵敏度高,可用于葡萄糖中糖类有关物质的检测。
参考文献
[ 1 ] 姚文兵.生物化学[M]. 8版.北京:人民卫生出版社,2016:6.
[ 2 ] 国家药典委员会.中华人民共和国药典:二部[S]. 2020年版.北京:中国医药科技出版社,2020:1514.
[ 3 ] European Pharmacopoeia Commission. European pharmacopoeia 9.0[S]. Strasbourg:European Drug Quality Administration,2017:2588-2590.
[ 4 ] The United States Pharmacopeial Convention. The United States pharmacopeia:40:volume 2[S]. Baltimore:United Book Press,2018:7677.
[ 5 ] Japanese Pharmacopoeia Committee. Japanese pharmacopoeia[S]. 17 version. Tokyo:Ministry of Health and Welfare of Japan,2017:988-989.
[ 6 ] 李毅群,王涛,郭书好.有机化学[M].北京:清华大学出版社,2007:387-401.
[ 7 ] 王婷,周欣蕊,林楠,等.高效液相色谱-蒸发光散射法检测发酵乳饮料中5种糖的含量[J].食品安全质量检测学报,2019,10(6):1542-1546.
[ 8 ] 洪小栩,石莹,宋雪洁,等.液相色谱柱进展及其在药品标准中的应用[J].药物分析杂志,2017,37(2):191-201.
[ 9 ] 林宏琳,陈华峰,林国斌.酰胺基亲水作用色谱-高效液相色谱-紫外法测定DHA颗粒中的磷脂酰丝氨酸[J].药物分析杂志,2017,37(11):1962-1966.
[10] 吴均成. HPLC-RID法测定灯盏花素葡萄糖注射液中葡萄糖的含量[J].北方药学,2018,17(8):5-7.
[11] 熊亚群,刘雁鸣.高效液相色譜技术在药用辅料检测中的应用新进展[J].中南药学,2015,11(1):61-64.
[12] 丁黎.药物色谱分析[M].北京:人民卫生出版社,2008:259-261.
[13] 仵淑红,王丽霞.高效液相色谱-示差折光检测器法测定牛磺酸颗粒和滴眼液中牛磺酸含量[J].中国药业,2018,27(17):29-31.
[14] 张红霞,张加余,刘颖,等.苦碟子注射液中果糖、葡萄糖和蔗糖的含量测定[J].中南药学,2014,12(4):366-369.
[15] 袁鹏,李蕊,杨瑞春.液相色谱-蒸发光散射检测法测定保健食品中8种糖的含量[J].实用预防医学,2020,27(9):1092-1095.
[16] 姚令文,刘瑞,聂黎行,等. UPLC-ELSD法快速测定生脉注射液中果糖、葡萄糖、蔗糖、麦芽糖含量[J].中国药事,2019,33(1):67-72.
[17] 王立萍,姚永清,梅芊,等. HPLC-ELSD法测定硫酸庆大霉素片的组分和有关物质[J].中国抗生素杂志,2019,44(5):558-562.
(收稿日期:2020-11-14 修回日期:2021-02-26)
(编辑:陈 宏)
