NH3在硼纳米管表面吸附的密度泛函理论研究
2021-07-11刘昌辉梁国俊李妍璐程秀凤
刘昌辉,梁国俊,李妍璐,程秀凤,赵 显
(1.山东大学晶体材料研究所,晶体材料国家重点实验室,济南250100;2.山东大学光学高等研究中心,青岛266237)
环境气体监测作为一个重要领域得到了人们的重视,许多研究都集中于开发合适的气敏材料,用于对超过规定浓度的有害化学气体进行连续监测和报警[1].众所周知,NH3是一种低沸点、易挥发、无色且具有腐蚀性的化合物[2],通常以气态形式存在,并具有特殊的刺激性气味,在低于37.95 mg/m3时也可以被觉察到[3],是大气中常见的含氮化合物.同时,NH3也可导致酸雨的形成.因此,在大气环境测量和控制中,对NH3气的有效监测和抑制提出了很高的要求.近年来,由于纳米管和纳米线等低维材料具有不同于体相材料独特的性质,使其在纳米器件中具有潜在的应用并备受关注.其中,碳纳米管由于在气体传感器等方面的重要应用而得到了广泛研究[4].然而,纳米技术的未来发展不应仅局限于碳的同素异形体,一些新兴的纳米管也被广泛用于气体监测中.Wu等[5]报道了NH3分子在纯氮化硼纳米管(BNNT)上的吸附性能和电子性质,表明NH3与BNNT的相互作用是物理吸附(吸附能为0.16 eV).Hadipour等[6]研究表明,氮化铝纳米管(AlNNT)也能够应用于NH3气的检测,由C,O和Si取代N原子的AlNNT对NH3气的吸附能力比AlNNT更加优异.与碳相比,硼是一个缺电子原子,其2s和2p轨道上有3个价电子,成键特性与碳明显不同[7].基于第一性原理的电子结构计算已从理论上预测了一系列新型硼基二维(2D)单层晶体结构[8,9].由于缺失电子的性质,由三角形和六边形孔洞基序共同组成的硼单层比仅有三角形或六边形孔洞基序的硼单层在能量上更稳定.目前,已报道的硼单层六边形孔洞密度(η)有1/4,1/5,1/6,1/7,1/8和1/9等几种情况[10].其中,α硼烯被预测为最稳定的结构[11],而实验上也在Cu,Ag等衬底上成功合成出了各种类型的硼烯结构,进一步证实了多种硼烯结构的存在[12~14].以碳纳米管的形成方式作为参照,硼纳米管(BNT)也可由硼烯通过不同卷曲方式来形成.如Liu等[15,16]利用密度泛函理论(DFT),通过卷曲两种不同类型的硼烯预测了一系列新的BNT.2004年,Ciuparu等[17]首次成功制备得到了纯硼单壁纳米管结构,表明了BNT的存在.
本文利用密度泛函理论,从微观角度研究了NH3在BNT表面的吸附过程,讨论了NH3的吸附位点、吸附能以及与BNT表面的相互作用,从理论上说明BNT作为NH3气气敏材料的潜在可能.
1 计算方法和模型
所有计算均采用第一性原理计算包ViennaAb-initioSimulation Package(VASP)[18]完成.采用投影缀加波方法(PAW)描述电子之间的相互作用[19],广义梯度近似(GGA)中的Perdew-Burke-Ernzerhof(PBE)来处理交换泛函[20].采用平面波作为基组,截断能为500 eV.第一布里渊区采用了(1×1×5)的k点网格,对晶胞和所有的原子位置都进行充分优化,直至力和能量的收敛标准分别小于0.2 eV/nm和10-4eV.所有的BNT沿管轴方向超胞到合适大小,长度在0.845~1.008 nm之间,BNT的直径在0.556~1.774 nm之间.沿管轴施加了一维(1D)周期性边界条件,并在另外两个垂直方向添加了1.5 nm的真空层.为了更好地描述BNT与NH3分子之间的相互作用,采用Grimme半经验DFT-D3方案进行色散校正[21].
与碳纳米管类似,BNT由通过将相应的硼片沿着手性矢量方向卷起得到.手性矢量用硼片晶格矢量表示为

式中:C为手性矢量;n和m为整数;a和b为晶格矢量.
对于由三角形和六边形孔洞混合而成的BNT结构,用η来区分不同BNT的类型,即六边形孔洞数量除以相应无孔三角形平面的原子数:

式中:Nholes为六边形孔洞的数量;Natom为实际平面结构的原子数.
采用聚合能(Ecoh)来阐明不同种类BNT的化学稳定性,Ecoh(eV)采用下式计算:

式中:Etot(eV)和Eat(eV)分别为整个系统的基态能量和一个孤立硼原子的基态能量;N为系统中的原子数.从这个定义可见,Ecoh的正值对应于稳定的结构.
采用吸附能(ΔE)来说明NH3分子在BNT上的吸附能力,ΔE采用下式计算:

式中:ΔE(eV)为吸附能;ENH3/BNT(eV)为NH3分子吸附在BNT表面后的总能量;ENH3(eV)和EBNT(eV)分别为单独的NH3分子和BNT的能量.根据定义,ΔE为负,表明吸附放热,绝对值越大,吸附作用越强.
差分电荷密度(Δρ)定义为分子在基体表面吸附前后的电荷密度之差,用于分析分子吸附前后的电子重排情况,计算如下:

式中:ρNH3/BNT为吸附体系总电荷密度;ρNH3和ρBNT分别为自由态NH3分子和BNT的电荷密度.

Fig.1 Atomic structures of boron sheets and armchair and zigzag boron nanotubes
在各种BNT结构中,选择最具代表性的α-BNT,β12-BNT,2-Pmmn-BNT和石墨型硼纳米管(g-B-BNT)4种BNT作为研究对象.同时,如图1所示,还考虑了扶手椅型(arm)和锯齿型(zz)两种BNT的结构以研究手性对BNT稳定性和吸附性能的影响.根据产生硼烯的种类和卷曲的方向,将4种BNT分别表示为α-BNT-arm和α-BNT-zz[图1(A)],β12-BNT-arm和β12-BNT-zz[图1(B)],2-Pmmn-BNT-arm和2-Pmmn-BNT-zz[图1(C)],g-B-BNT-arm和g-B-BNT-zz[图1(D)].
2 结果与讨论
2.1 BNT结构的稳定性
BNT具有良好的稳定性是可吸附NH3分子的前提.因此,首先通过计算各种类型BNT的Ecoh来对比它们的相对稳定性,以此来寻找最为合适的吸附基体.图2(A)给出了4种具有不同手性的BNT的Ecoh随直径的变化.可见,在直径为0.500~1.800 nm范围内,它们的相对稳定性顺序为α-BNT>β12-BNT>2-Pmmn-BNT>g-B-BNT,与文献[22]相符.除了2-Pmmn-BNT以外,手性对纳米管的稳定性影响很小,说明采用不同方式对单层硼烯卷曲不会明显降低BNT的稳定性,这也是实验成功合成多种类型BNT的基础.而对于2-Pmmn-BNT-arm和2-Pmmn-BNF-zz而言,二者的相对稳定性差异较大,主要因为2-Pmmn硼纳米管的σ键具有方向性,使得该类型纳米管在不同方向上卷曲的难易程度不同:沿着扶手椅型方向卷曲时,强大的σ键使得平面不易弯曲,聚合能低;沿着锯齿型方向卷曲时,由于不存在σ键的阻碍作用,聚合能则较高,结构更稳定.此外,还可以看出,随着BNT直径的增加,不同纳米管的稳定性具有相同的趋势,当纳米管直径大于一定尺寸时,纳米管的稳定性与自身对应的片状硼烯结构非常接近.表明对于大尺寸BNT,量子约束效应和曲率效应很小,而且纳米管的物理性质与单层硼烯的性质非常相似,因此可以用单层硼烯来定性类比BNT的性质.如Bezugly等[23]对比了实验得到的大直径BNT、纳米线和理论计算的α硼烯的功函数,发现三者具有很好的一致性.正是基于此,计算了二维α硼烯的声子谱[图2(B)],以证明硼原子之间化学成键形成单层结构的动力学稳定性.可见,二维α硼烯的声子谱在整个布里渊区没有虚频,能够证明其动力学稳定性,也表明由该硼烯结构卷曲形成的α-BNT在动力学上也是稳定的.

Fig.2 Cohesive energies of investigated armchair(arm)and zigzag(zz)boron nanotubes(A)and phonon spectrum structures ofα-sheet boronene(B)
2.2 NH3在BNT表面的吸附
基于稳定性的考虑,选择最稳定的α-BNT作为NH3的吸附载体.原始α硼烯及对应的α-BNT-arm和α-BNT-zz的几何结构如图1(A)所示.可见,优化后的自由态硼烯保持了完美的单层结构特征,硼烯上的原子通过B—B键键合,键长分布在0.167~0.169 nm之间,平均为0.168 nm,形成以三角形和六边形孔洞为基序独特的平面结构,每个占据中心的六边形彼此隔离,六边形孔洞密度值η为1/9.在α硼烯中,硼原子具有两种配位数(CN),分别为CN5和CN6.由自适应自然密度划分方法[24]分析得出,这两种配位数的硼原子具有不同的电子特性.CN5硼原子是由2个三中心两电子(3c-2e)σ键和一个四中心两电子(4c-2e)σ键连接的,而CN6硼原子由离域的π键连接,两种不同的键合方式说明它们自身对分子的吸附具有不同的响应.然而,与平面硼烯不同,α-BNT的原子不再保持同一个平面之中,相邻的两个CN6位点硼原子其中一个会向着管内凹陷.这种凹陷会随着直径的增加有所减缓,同时,凹陷程度也因不同的手性有所不同.
根据不同的原子配位数和凹陷情况,选取4个顶(Top)位点作为NH3吸附的第一潜在位点,同时选取不同的桥位(B)、孔位(H)和三中心键位(TC)作为NH3吸附的第二潜在位点来研究NH3在α-BNT表面的吸附情况,首先对第一潜在位点的4个Top位(CN51,CN52,CN61和CN62)进行NH3吸附.相应的4个位点及吸附能如图3所示,由于卷曲的作用,CN62位点的B原子会沿着直径方向向管内凹陷,BNT-arm要比BNT-zz的凹陷程度严重.而CN61位点的B原子在不同手性情况下均保持和整个管壁平面一致.两个相邻配位数为5的B原子(CN51和CN52)虽然保持在同一平面且具有一致的对称性,但由于受到周围两个CN6位点B原子的影响,其对NH3的吸附具有差异性.从图3(C)和(D)可见,4个位点上NH3的吸附能均为负值,说明NH3与α-BNT的4种Top位点有较强的相互作用,为化学吸附.在这些Top位点上,NH3均保持了自由态分子的三角锥空间构型,但由于吸附时B,N原子的相互作用,使得不同位点的B原子微微向着管外突出.由于CN62位点的B原子本来是内凹的,N原子的作用使得凹陷程度减少,甚至向外突出.而NH3分子的键角也因相互作用而从原始的107°增大到108.39°左右.此外,B—N键长在不同手性和位点情况下几乎保持一定,大约为0.164 nm,与BN晶体中B—N键长0.157 nm相符,表明B,N之间形成了共价键,这证明了NH3与BNT之间为化学吸附.对于同一个位点,随着直径(手性数)的增加,NH3吸附能具有不同的变化趋势.对于α-BNT-arm[图3(A)和(C)],各个位点和直径呈线性变化关系,除了CN52位点,不同位点的吸附能随着直径的增加而增强.而对于α-BNT-zz[图3(B)和(D)],随着直径的增加,各个位点的吸附能均呈先减小后增大的趋势.表明尺寸效应对NH3吸附的影响不是简单的BNT尺寸越小吸附能力越好的关系,而是存在一个最适合NH3分子吸附的直径.对于α-BNT-zz而言,这个最优的“拐点”处于手性为(4.4)和(5.5)之间,即直径在1.100~1.400 nm之间.而对于α-BNT-arm而言,最优的“拐点”显然不在1.100~1.400 nm直径范围内,说明不可用常规的尺寸效应来寻找最优的吸附直径.然而,尺寸效应对NH3分子的吸附构型的影响却很小,在不同的直径下,NH3总是保持着正对B原子,使得NH3分子的各个键角相等.同样,NH3分子构型在不同的配位数位点也基本保持不变.然而,对于不同的配位数位点,NH3的吸附能具有很大的变化,相对于CN5位点,CN6位点具有更负的吸附能,表明NH3分子会更加优先吸附在CN6位点,也说明由于凹陷引起的位点环境变化导致的对NH3吸附的影响要小于因不同配位数的自身电子特性不同而产生的影响.然而,凹陷的程度会对相同配位数的不同位点有较大的影响.由于(6.0)α-BNT-arm相对于(10.0)α-BNT-arm直径更小,原子下凹程度更大,所以相对于(6.0)α-BNT-arm,CN61和CN62的吸附能相差更大.而随着直径的增加,这一差距逐渐减小,对于CN51和CN52位点具有相似的结果.相比于α-BNT-arm,α-BNT-zz原子凹陷程度低,且随直径变化小,所以对于相同配位的两个位点,NH3的吸附能差距小,变化缓慢.除了Top位点,桥位和孔位等都有可能是NH3的吸附位点,因此还对这些第二潜在吸附位点进行了NH3分子吸附的研究.计算表明,在六边形孔洞边缘的桥位,NH3分子趋向偏移吸附到周围的CN5顶位点,而非边缘的桥位点以及孔洞中心位点均不能有效吸附NH3分子.此结果在不同手性、不同直径的BNT具有一致性,进一步说明在一定范围的管径内,尺寸效应和手性无法从根本上改变位点的吸附性能.

Fig.3 Adsorption sites(A,B)and corresponding adsorption energy(C,D)of NH3 on armchair(A,C)and zigzag(B,D)boron nanotubes
为了进一步了解配位数对NH3吸附的影响,分别以直径相近的(8.0)α-BNT-arm和(5.5)α-BNT-zz为基础构建了含有单空位缺陷的α-BNT,并产生了配位数为4的新位点.如图4(A)和(B)所示,依次对两种BNT的CN5位点和CN6位点进行B缺陷处理(红色小球代表缺失的B原子),可以分别得到两种不同的缺陷构型.因为手性和空位原子的不同,不同的CN4位点在键合方式和空间位置有所不同,分别对不同的CN4位点进行NH3吸附能的计算[图4(C)].可见,无论是(5.5)α-BNT-zz还是(8.0)α-BNTarm,CN41位点的吸附能均比CN42和CN43位点吸附能高一倍左右,说明对于同一手性BNT的不同位点,NH3的吸附能差别较大,表明不同的键合方式直接影响了NH3的吸附;而NH3分子分别在(5.5)α-BNT和(8.0)α-BNT的相同位点表现出十分相近的吸附能,说明手性引起的位点空间位置的变化对NH3仅产生很小的影响.因此,得到了与NH3分子在完整BNT表面吸附相似的结果:在相近的直径下,手性对NH3的吸附影响可以忽略,而吸附位点的键合方式是影响NH3吸附强弱的关键因素.此外,还计算了缺陷α-BNT的第二潜在吸附位点的吸附行为,桥位和孔位依旧不能有效地进行NH3的吸附.
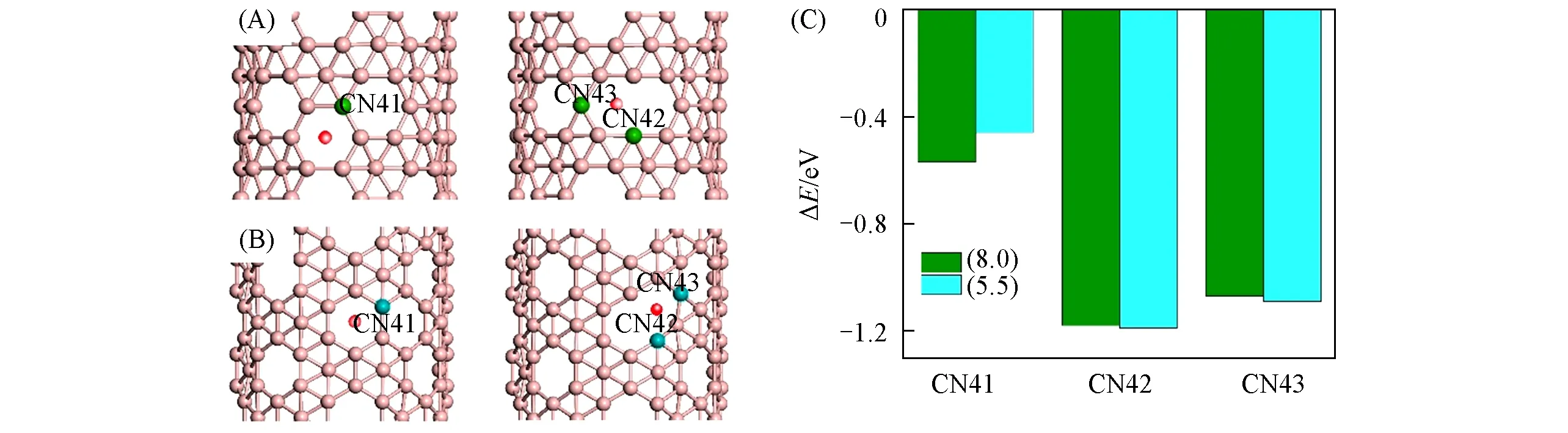
Fig.4 Adsorption sites(A,B)and corresponding adsorption energy(C)of NH3 on(8.0)α-BNT-arm(A,C)and(5.5)α-BNT-zz(B,C)containing different sites of boron vacancy
2.3 电子性质
为了探究吸附NH3分子对BNT电子结构的影响,首先选取(8.0)α-BNT-arm和(5.5)α-BNT-zz两种BNT,分别计算得到纯相BNT的能带结构图以及在不同位点吸附了NH3分子的能带结构,如图5所示.可见,原始的(8.0)α-BNT-arm具有明显的间接带隙,约为0.32 eV,是半导体性的,是原子下凹产生的结果.而在原始的(5.5)α-BNT-zz的能带图上,有一条能带穿过费米能级,表明该结构是金属性的.两种手性的α-BNT具有不同的能带结构主要是由原子下凹程度不同造成的.(8.0)α-BNT-arm的CN61位点B原子相比于(5.5)α-BNT-zz下凹程度更明显,因此具有更明显的导体到半导体的转变特征.然而,当NH3分子吸附在两种不同的α-BNT上时,能带结构却有着不同的变化趋势.对于(8.0)α-BNT-arm,当NH3吸附在CN51和CN62位点时,带隙趋近减小,特别是NH3在CN51吸附后体系转变为导体性质;而吸附在CN52和CN61位点其带隙几乎保持不变.这种不同的电子结构特征是因为NH3分子在CN51位点的吸附作用使得基体的下凹程度减小,而在CN52和CN61位点吸附的小分子对基体几何结构影响很小,因此保持了原始的性质.当NH3分子吸附在(5.5)α-BNT-zz不同位点时,带隙有打开的趋势,也与吸附后基体的几何结构变化有关.

Fig.5 Electronic band structures for pure(8.0)α-BNT-arm(A1)and(5.5)α-BNT-zz(B1),as well as the corresponding BNTs adsorbed NH3 at CN51(A2,B2),CN52(A3,B3),CN61(A4,B4)and CN62(A5,B5)sites of(8.0)α-BNT-arm(A2—A5)and(5.5)α-BNT-zz(B2—B5)
采用Bader电荷分析[25]结合电荷差分密度来反映NH3分子在BNT表面吸附前后的电子分布状态以及电荷转移情况.NH3分子在吸附后转移的电荷量越多,说明其与BNT表面相互作用越强,吸附能越大.在自由态的NH3分子中,N原子具有较强的电负性,而整个分子为电中性.由于NH3分子和BNT表面的相互作用,整个分子转移电子数随着吸附环境而改变,因而产生了各不相同的吸附能力.计算了从BNT到NH3分子的电子转移数(CT)随着BNT直径和吸附位点的变化,结果列于表1和表2.从表1可见,在一定的直径范围内,NH3趋向于失电子.这种由尺寸效应主导的电子转移和手性几乎没有关系,如对于(7.0)和(4.4),(8.0)和(5.5)以及(10.0)和(8.0)BNT,它们具有相近的直径,而吸附在CN61位点的NH3向BNT的电子转移数基本一致,与吸附能和手性之间的弱相关性结果一致.而随着直径的增加,尺寸效应减弱,电子转移的数量也有所减少.而忽略尺寸效应的影响,在同一直径BNT的不同位点进行吸附,配位数越大,电子转移数越多.由表2可见,CN6位点相比于CN5位点转移的电子数更多,直接导致了CN6位点在整体吸附性能上要优于CN5位点.为了直观地显示电子转移及成键情况,分别对(8.0)α-BNT-arm和(5.5)α-BNT-zz的4个NH3吸附位点做了差分电荷图(图6).可见,靠近BNT表面的N原子损失了一些电子,而在靠近NH3分子的B原子上方有明显的电子积聚,表明N—B键发生了较强的相互作用,因此NH3能在α-BNT表面进行有效的吸附.

Table 1 Electron transfer number(CT)from BNT to NH3 varing with different chirals at adsorption site CN61

Table 2 Electron transfer number(CT)from BNT to NH3 varing with different sites under the chiral of(6.0)and(3.3)
Fig.6 Charge density differences of NH3 on different sites of(8.0)α-BNT-arm(A)and(5.5)α-BNT-zz(B)
3 结 论
通过密度泛函理论研究了NH3分子在α-BNT上的吸附行为.计算结果表明,NH3分子在不同直径和手性的α-BNT表面均具有负的吸附能,可见二者是化学吸附,因此α-BNT是潜在的NH3气敏材料.然而,NH3在BNT表面的吸附能随着纳米管的手性和直径发生变化:(1)在一定直径下,不同手性的纳米管对NH3吸附能仅有很小的影响,这是因为手性的改变仅会影响α-BNT原子的凹陷程度和位点的空间位置,进而产生微小的作用;(2)在一定的直径范围内,在BNT不同位点吸附NH3其吸附能具有不同的变化趋势,表明吸附能不仅是由尺寸效应决定,而是由位点自身电子特性、尺寸和手性等多因素综合决定的.进一步通过电子结构分析可以得出,NH3分子通过改变基体几何结构而改变整个体系的电子结构,NH3在不同位点的有效吸附是通过B—N键较强的相互作用而产生化学吸附的结果,在同样产生化学吸附的前提下,不同吸附位点对体系电子结构的影响基本可以忽略.
