高表达CXCR4可通过乳酸脱氢酶A磷酸化诱导肺腺癌细胞产生吉非替尼耐药
2021-07-03祝巧良卢春来
祝巧良, 卢春来, 古 杰, 葛 棣
复旦大学附属中山医院胸外科,上海 200032
肺癌已成为全球范围内发病率和死亡率最高的恶性肿瘤[1]。近年来,肺癌的病理类型谱发生了显著变化:肺腺癌的发生率不断上升,已超越鳞癌,成为最常见的肺癌组织学类型[1]。在肺腺癌中,表皮生长因子受体(epidermal growth factor receptor,EGFR)的激活突变是最常见的驱动基因突变,以吉非替尼和厄洛替尼为代表的EGFR酪氨酸激酶抑制剂(tyrosine kinase inhibitors,TKIs)可显著延长含EGFR敏感突变(19del、21-L858R)肺腺癌患者的无进展生存期(progression-free-survival,PFS)[2]。但是,即使对EGFR-TKIs初治高度敏感的患者,在经过9~11个月的中位PFS后,几乎都不可避免地产生耐药,这极大限制了其临床治疗效果[3]。目前关于EGFR-TKIs的耐药机制主要包括:靶基因的二次突变,如T790M突变[4];EGFR旁路的持续激活,如MET扩增[5];表型和组织学类型的转化,如小细胞转化和上皮间质转化(epithelial to mesenchymal transition, EMT)[6],但仍有部分耐药机制尚未阐明。因此,深入研究EGFR-TKIs耐药的相关机制,制订相应的策略,具有重要的理论价值和临床意义。
CXC趋化因子受体4(CXCR4)在多种肿瘤细胞中明显高表达,与肿瘤的发生和发展密切相关[7-8]。且CXCR4的高表达可显著降低肿瘤的化疗药物敏感性、诱导化疗耐药[9-10]。值得注意的是,肺腺癌细胞在出现吉非替尼耐药后,CXCR4的表达水平较耐药前显著升高[11]。但CXCR4高表达与肺腺癌细胞吉非替尼耐药的相关性目前仍不明确。因此,本研究旨在通过体外及体外试验初步探讨CXCR4高表达诱导肺腺癌细胞产生吉非替尼耐药的相关机制。
1 材料与方法
1.1 细胞培养 PC9(含EGFR-19del突变的吉非替尼敏感的肺腺癌细胞)、PC9/GR(含 EGFR-19del突变的吉非替尼耐药的肺腺癌细胞)均购于湖南丰晖生物科技有限公司;PC9细胞培养于含10%胎牛血清的DMEM培养液中,PC9/GR细胞按细胞说明书要求培养于加有2 μmol/L吉非替尼(Selleck公司)的含10%胎牛血清的DMEM培养液中,置于37℃、5%CO2的恒温细胞培养箱中培养。
1.2 RNA干扰 PC9/GR细胞对数生长期时,消化后铺板,待细胞融合度达到30%~50%时,采用脂质体转染方法对细胞进行siRNA转染以下调胞内CXCR4水平,转染方法按照Lipo2000说明书。针对CXCR4的mRNA设计siRNA:siCXCR4-1的靶序列为5′-GGU ACU UUG GGA ACU UCC U-3′(F)、5′-AGG AAG UUC CCA AAG UAC C-3′(R);si-CXCR4-2的靶序列为5′-CCU GUC CUG CUA UUG CAU U-3′(F)、5′-AAU GCA AUA GCA GGA CAG G-3′ (R);siCXCR4-3的靶序列为5′-GUG AGU UUG AGA ACA CUG U-3′(F)、5′-ACA GUG UUC UCA AAC UCA C-3′(R)。siRNA-NC为非特异性非相关的RNA片段,作为对照组。
1.3 慢病毒感染 采用CRISPR/CAS9(汉尹生物科技有限公司)技术设计、构建、包装过表达ovCXCR4及对照ovCtrl的慢病毒载体,测定慢病毒滴度;2×105/孔的PC9细胞接种于6孔板,培养24 h后,移入生物安全柜操作,按照产品说明书滴加慢病毒;按1∶1 000比例加入polebrene混匀;慢病毒感染24 h后换液;48 h后加入适量嘌呤霉素继续培养72 h,去除未成功感染慢病毒的细胞;待细胞扩增至长满80%的6孔板后,消化、收集细胞。qRT-PCR、Western印迹检测CXCR4表达情况,鉴定感染效果。
1.4 qRT-PCR和Western印迹检测 细胞总RNA提取、qRT-PCR、细胞总蛋白提取、Western印迹检测等步骤参照既往文献[12]报道方法进行。细胞总RNA提取、qRT-PCR:吸去培养液,PBS清洗1次,加入Buffer RZ裂解液裂解细胞,按细胞总RNA提取试剂盒(天根生化科技有限公司)说明书提取细胞总RNA,经反转录获得cDNA,将cDNA用作RT-PCR模板,按TaKaRa荧光定量PCR试剂盒说明书配制试剂和样品,加入20 μL反应体系于ABI Prism 7500实时PCR仪器反应。CXCR4引物合成于上海生工生物科技有限公司,为5′-TGT CAT CTA CAC AGT CAA CCT C-3′(F)、5′-CAA CAT AGA CCA CCT TTT CAG C-3′(R)。β-actin引物序列为5′-TGA CGT GGA CAT CCG CAA AG-3′(F)、5′-CTG GAA GGT GGA CAG CGA GG-3′(R)。
细胞总蛋白提取、Western印迹检测:去细胞培养液,PBS洗涤2遍,加入RIPA裂解液冰上裂解细胞10 min,4℃离心(13.8×g)10 min后取上清液,获得总蛋白。BCA法测定蛋白浓度;加入SDS加样缓冲液,100℃煮沸10 min使蛋白变性。按5~10 g样品上样,经电泳、转膜、封闭、一抗孵育过夜、洗膜、二抗孵育、洗膜后,采用ECL试剂显色发光。一抗货号:pEGFR(#3777), EGFR(#2085), pAKT(#2965), AKT(#9272), pERK1/2(#4370), ERK1/2(#4695), Cleaved caspase 3(#9664), BAK(#6947), BCL-2(#3498), BCL-XL(#2764), 乳酸脱氢酶A(lactate dehydrogenase A,LDHA,#3582),pLDHATyr10(#8176), β-actin(#4970)均购自美国赛信通公司;CXCR4抗体(ab181020)购自英国Abcam公司。
1.5 克隆形成、CCK-8、流式细胞凋亡术
1.5.1 克隆形成实验 将细胞按1.5×103个/孔接种到6孔板中,细胞贴壁24 h, 根据实验目的(转染siRNA/加入抑制剂)经相应处理后持续培养2周,4%多聚甲醛固定、结晶紫染色,显微镜下随机选取5个视野中含50个以上细胞的集落,计数、求平均值并统计。
1.5.2 CCK-8检测 将细胞按5×103个/孔接种到96孔板中,细胞贴壁24 h,根据实验目的(转染siRNA/加入抑制剂)经相应处理72 h后,进行细胞活力检测,具体步骤按照CCK-8说明书进行;将CCK-8试剂与无血清培养液进行1∶9混合,弃去细胞培养液,分别于对应孔中加入100 μL CCK-8混合液,37℃孵育1~2 h,采用酶标仪检测450 nm的光密度(D)值,计算并统计细胞相对增殖活力。
1.5.3 流式细胞凋亡术 将细胞按4×104个/孔接种到24孔板中,细胞贴壁24 h,根据实验目的(siRNA干扰/加入抑制剂)经相应处理48 h后,消化、收集细胞。4℃预冷的PBS洗涤细胞,2次4℃离心(300×g)5 min;250 μL 1×binding buffer重悬细胞;根据Annexin Ⅴ-FITC/PI细胞凋亡检测试剂盒(翌圣生物科技有限公司)说明书,取100 μL细胞悬液,加入流式抗体5 μL APC-Annexin Ⅴ和10 μL PE-PI,室温避光反应15 min,每管加入4℃预冷的PBS 400 μL终止反应,上机并检测[13]。
1.6 裸鼠皮下成瘤 动物研究和实验方案已获得复旦大学实验动物中心批准(B2021-128),根据动物安置和护理准则对动物进行饲养和处理。本研究使用4~6周龄清洁级雌性裸鼠(上海杰思捷公司),饲养于复旦大学上海医学院SPF级动物房(12 h/12 h光照/黑暗周期)。每只小鼠皮下注射2×106个细胞,荷瘤小鼠饲养2周后随机分组(3只小鼠/组):Ctrl(PBS)、Gef (gefitinib;30 mg/kg,灌胃,qd×3周);AMD(AMD 3100; 5 mg/kg,腹腔注射,qd×4周);AMD+Gef(AMD预处理1周,然后用Gef处理);Oxa(oxamate ;750 mg/kg,腹腔注射,qd×4周);Oxa+Gef(Oxa预处理1周,然后用Gef处理)。每3 d用游标卡尺测量皮下瘤直径,评估并统计。
1.7 免疫共沉淀 扩增并收集过表达CXCR4的PC9细胞,细胞经IP裂解液裂解,取上清后加入抗CXCR4抗体/IgG,4 ℃振荡过夜,免疫复合物溶液与Protein A/G磁珠在室温下混合孵育,洗涤,留取阴性参照、阳性参照、IP液,经上样、电泳、考马斯亮蓝染色后送上海维基生物科技有限公司行蛋白胶条Shotgun质谱分析,Western印迹验证质谱。具体步骤参照既往文献[14]。
1.8 葡萄糖消耗量和乳酸生成量测定 将细胞按3×105个/孔接种到6孔板中,贴壁24 h;将培养液更换为无酚红(赛默飞世尔科技有限公司,美国)低葡萄糖DMEM液。根据实验目的(siRNA干扰/加入抑制剂)经相应处理48 h后,从每个孔中收集50 μL培养液等分试样,以950 μL蒸馏水(1∶20)稀释。使用葡萄糖检测试剂盒(Sigma-Aldrich),按照产品说明书设定空白孔、标准孔、测定孔,加入显色剂后37℃环境反应15 min,酶标仪测定505 nm处的各测试孔D值,计算并统计细胞的葡萄糖消耗量;使用乳酸检测试剂盒(南京建成生物工程公司),按照产品说明书设定空白孔、标准孔、测定孔,加入显色剂后37℃环境反应10 min,酶标仪测定530 nm处的各测试孔D值,计算并统计细胞的乳酸生成量。具体步骤参照既往文献[15]。
1.9 统计学处理 采用GraphPad Prism软件分析数据,采用t检验比较差异。细胞实验每项重复3次。检验水准(α)为0.05。
2 结 果
2.1 PC9/GR细胞的吉非替尼耐药性鉴定 克隆形成(图1A)、CCK-8检测(图1B)结果显示,在吉非替尼作用下,PC9/GR显示出显著增强的增殖能力;PC9/GR、PC9细胞的吉非替尼半抑制浓度IC50分别是19.15 μmol/L、0.195 μmol/L,前者明显高于后者;在吉非替尼作用下,PC9细胞中pAKT、pERK明显下调,而在PC9/GR细胞中无明显下调,提示PC9/GR可显著抵抗吉非替尼对EGFR下游通路的抑制作用(图1C);PC9/GR较PC9细胞存在显著增强的抗凋亡能力(图1D);PC9细胞的促凋亡蛋白如cleaved-caspase3、BAK明显上调而抗凋亡蛋白如BCL-XL、BCL-2明显下调,PC9/GR中凋亡蛋白均变化不明显(图1E)。体内实验提示,吉非替尼可显著抑制PC9细胞的皮下成瘤,而对PC9/GR的抑制并不明显(图1F)。以上结果表明PC9/GR细胞存在良好的吉非替尼耐药性。qRT-PCR和Western 印迹检测结果(图1G)显示,CXCR4在PC9/GR细胞中的表达明显高于PC9细胞。
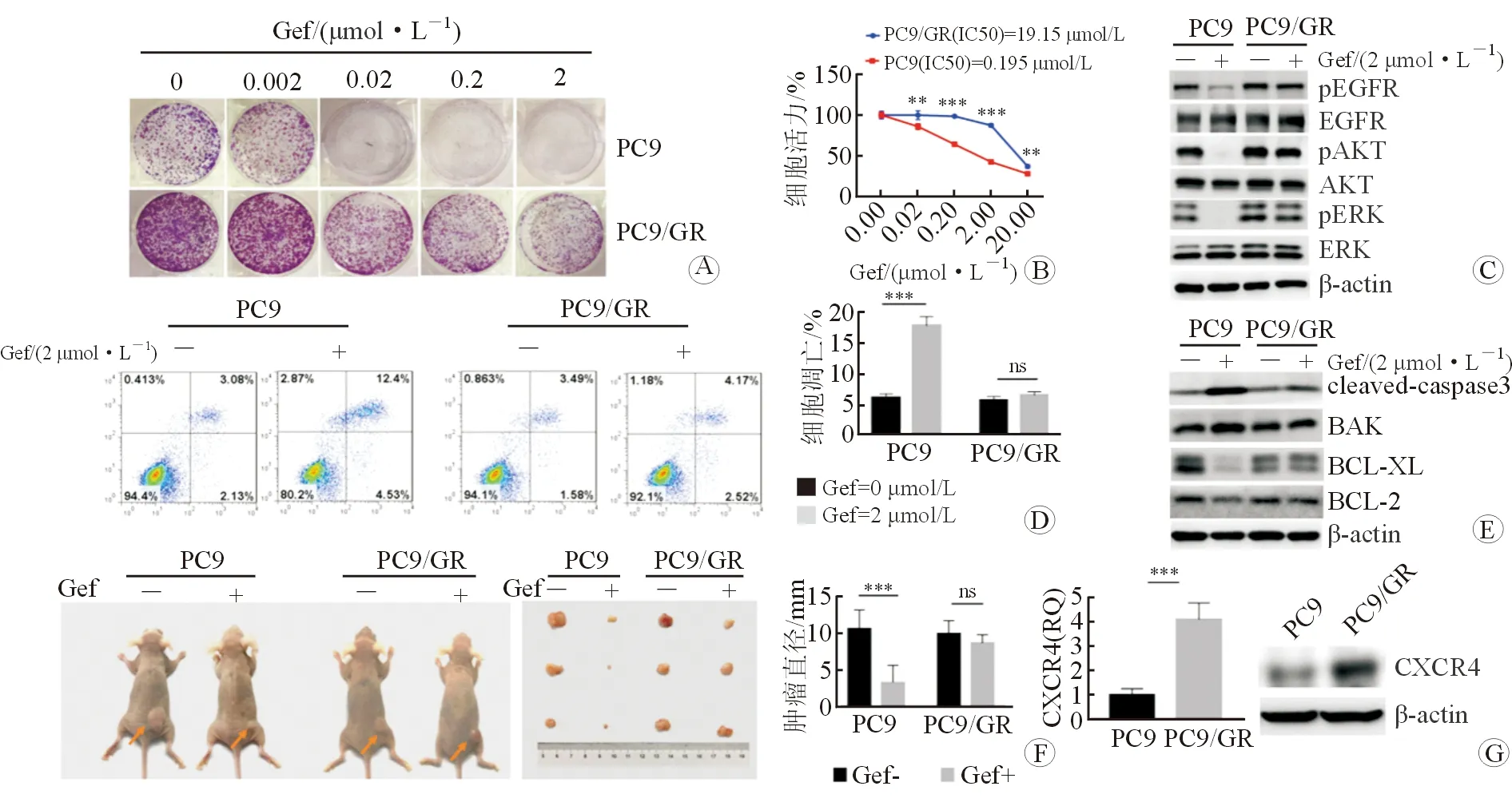
图1 PC9/GR细胞的吉非替尼耐药性鉴定
2.2 抑制CXCR4对PC9/GR细胞耐药的调控 结果(图2)显示:抑制PC9/GR细胞的CXCR4后,吉非替尼可显著抑制细胞克隆形成(图2A);降低吉非替尼IC50(图2B);促进细胞凋亡(图2C),上调促凋亡蛋白、下调抑凋亡蛋白(图2D),抑制细胞的EGFR下游信号通路(图2E);在体内,吉非替尼可显著抑制细胞的皮下成瘤(图2F)。以上结果表明,CXCR4受抑后可显著逆转PC9/GR细胞的吉非替尼耐药性。
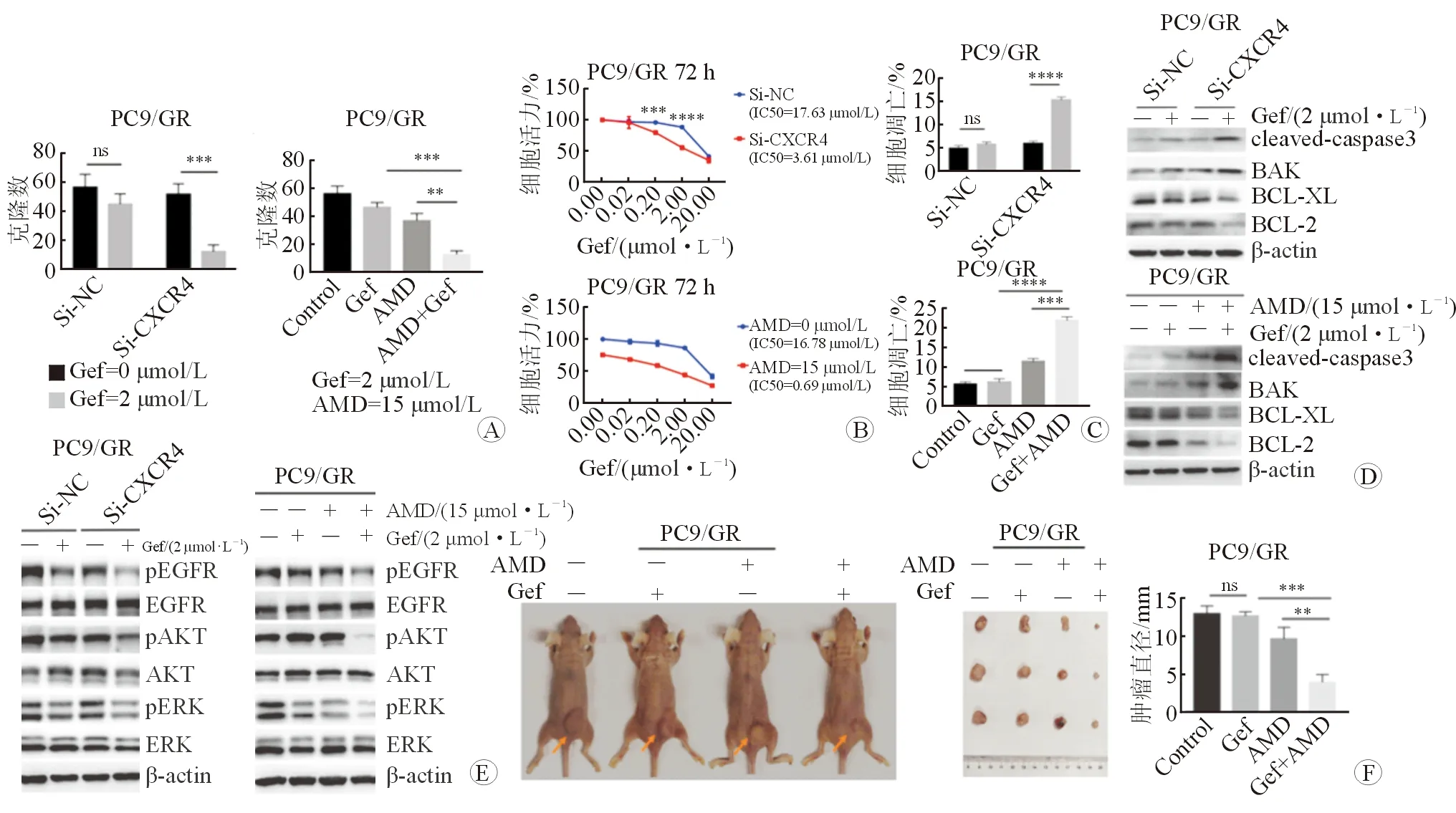
图2 抑制CXCR4可逆转PC9/GR细胞的吉非替尼耐药性
2.3 CXCR4过表达对PC9细胞耐药的调控 过表达CXCR4后,PC9细胞的克隆形成能力明显增强(图3A);吉非替尼IC50显著增加(图3B);抗凋亡能力明显增强(图3C、图3D);显著抵抗吉非替尼对细胞EGFR下游通路的抑制作用(图3E);在体内,CXCR4过表达细胞可显著抵抗吉非替尼对皮下成瘤的抑制(图3F)。以上结果表明,过表达CXCR4可诱导PC9细胞产生吉非替尼耐药。

图3 过表达CXCR4可诱导PC9细胞产生吉非替尼耐药
2.4 CXCR4过表达诱导PC9细胞耐药的机制 结果(图4)显示:多种蛋白可直接结合于CXCR4分子,其中LDHA与CXCR4的结合丰度最高(图4A); CXCR4抗体可检测到结合的LDHA(图4B),而LDHA抗体可检测到结合的CXCR4(图4C)。因此,LDHA和CXCR4存在相互作用。在PC9细胞中过表达CXCR4时,LDHA蛋白水平没有改变,而LDHA的Y10磷酸化水平显著增加(图4D);细胞的葡萄糖消耗量(图4E)和乳酸生成量明显上调(图4F)。因此,CXCR4过表达可促进LDHA磷酸化,从而增强细胞的有氧糖酵解。
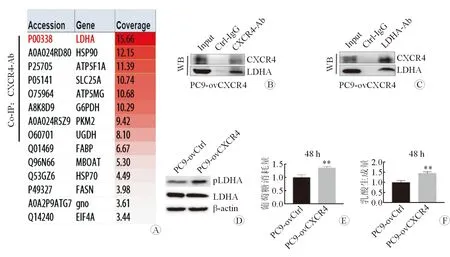
图4 CXCR4过表达促进LDHA磷酸化并增强PC9细胞的有氧糖酵解
2.5 抑制CXCR4对PC9/GR细胞有氧糖酵解的调控 结果显示:PC9/GR细胞中的pLDHA水平显著高于PC9细胞(图5A),葡萄糖消耗量(图5B)和乳酸生成量(图5C)明显升高。抑制CXCR4不改变LDHA表达,但显著下调了PC9/GR细胞中的pLDHA水平(图5D),并明显降低PC9/GR细胞的葡萄糖消耗量(图5E)和乳酸生成量(图5F)。因此,抑制CXCR4可下调PC9/GR细胞的LDHA磷酸化并降低其有氧糖酵解功能。

图5 抑制CXCR4可下调PC9/GR细胞中LDHA磷酸化并降低有氧糖酵解
2.6 有氧糖酵解对细胞耐药的影响 以糖酵解抑制剂(Oxamate和2-DG)预处理过表达CXCR4的PC9细胞,结果发现:吉非替尼可显著抑制细胞增殖(图6A);明显降低细胞的吉非替尼IC50(图6B);促进细胞凋亡(图6C),上调促凋亡蛋白、下调抑凋亡蛋白(图6E);在体内,吉非替尼可显著抑制细胞的皮下成瘤(图6D)。因此,抑制有氧糖酵解可逆转CXCR4诱导的吉非替尼耐药。
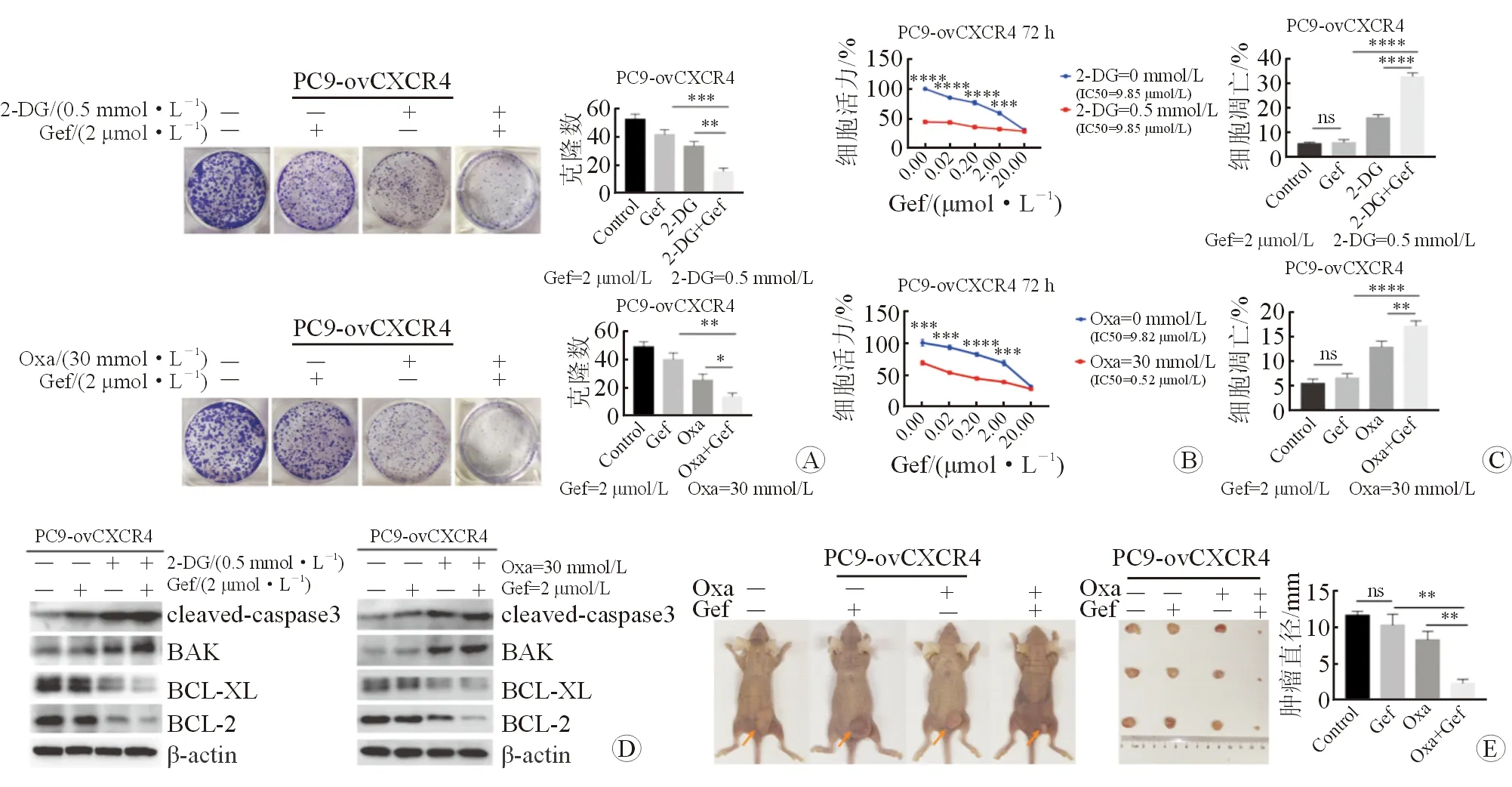
图6 抑制有氧糖酵解可逆转CXCR4诱导的吉非替尼耐药
3 讨 论
在过去的十几年中,EGFR-TKIs使含EGFR敏感突变的非小细胞肺癌明显受益。但是,EGFR-TKIs的临床获益是有限的:几乎所有患者都不可避免地出现了疾病的进展,此即EGFR-TKIs耐药,也给肺癌的临床治疗带来了新的困难[16-18]。因此,深入研究EGFR-TKIs的耐药机制,有针对性地制订相应策略,对控制病程、改善预后具有重要的理论意义和临床应用价值。
CXCR4作为一种7次跨膜的G蛋白偶联受体,可异常表达于多种肿瘤细胞中,在与其配体SDF-1α结合后,酪氨酸激酶活性被激活,从而引发下游信号蛋白的酪氨酸磷酸化,促进肿瘤细胞的增殖、迁移和转移。Phillips等[19]报道,多种非小细胞肺癌细胞系中存在CXCR4高表达;干扰细胞的CXCR4表达或使用特异性抑制剂阻断CXCR4通路后可明显降低细胞的侵袭和迁移能力。此外,CXCR4高表达可明显促进肺癌细胞的淋巴结转移,且与患者的总体预后负相关[20]。随着研究的深入,CXCR4过表达可诱导肿瘤细胞产生化疗耐药,阻断CXCR4通路后可恢复癌细胞的化疗敏感性[9-10]。引起我们关注的是,据Jung等[11]报道,野生型EGFR(A549/GR)的肺腺癌细胞在出现吉非替尼耐药后,CXCR4的表达水平较母细胞(A549)显著升高。但CXCR4是否参与了吉非替尼耐药,尚无相关报道。本研究证实,PC9/GR细胞的吉非替尼耐药性良好,且存在CXCR4明显高表达,CXCR4受抑后可逆转PC9/GR的吉非替尼耐药;而在PC9细胞中过表达CXCR4后可诱导细胞产生吉非替尼耐药。本研究首次揭示了在含EGFR敏感突变的肺腺癌细胞中,CXCR4的表达和活性对吉非替尼的药物敏感性具有重要的调控作用。
与正常细胞在有氧条件下使用线粒体氧化磷酸化供能方式不同,肿瘤细胞即使在氧供充足的情况下亦优先通过葡萄糖的糖酵解供能,即有氧糖酵解,以维持肿瘤细胞的无限增殖和持续生长,这种能量重编被称为Warburg效应[21]。作为有氧糖酵解的关键酶之一,LDHA可催化丙酮酸和NADH转化为乳酸和NAD+,其异常表达与多种恶性肿瘤密切相关。本研究证实,CXCR4不影响LDHA蛋白的表达水平;但是过表达CXCR4可显著促进细胞的LDHA磷酸化,上调有氧糖酵解水平;抑制CXCR4后细胞中LDHA磷酸化和糖酵解水平均显著下调;抑制有氧糖酵解可逆转CXCR4过表达所诱导的肺腺癌细胞吉非替尼耐药。
本研究尚有不足之处:CXCR4是否同样在含EGFR突变的其他细胞系,如HCC827、H1975等细胞中,发挥吉非替尼耐药的调控作用需要进一步验证;另外,还需收集临床肺腺癌肿瘤样本,检测吉非替尼耐药前后的CXCR4和LDHA、pLDHA水平,进一步验证CXCR4和LDHA的相关性。
综上所述,本研究结果表明CXCR4过表达可诱导肺腺癌细胞产生吉非替尼耐药;从机制上揭示了CXCR4过表达通过对LDHA磷酸化的促进而增强细胞有氧糖酵解功能;有氧糖酵解受抑后可显著逆转CXCR4诱导的吉非替尼耐药。本研究为解释吉非替尼耐药的发生机制补充了理论基础,对克服吉非替尼耐药提供了新的靶点和实验依据。
