Enhancer of zeste homolog 2 contributes to apoptosis by inactivating janus kinase 2 / signal transducer and activator of transcription signaling in inflammatory bowel disease
2021-06-24JieZhouYangYangYiLingWangYueZhaoWenJingYeSiYaoDengJinYiLangShunLu
Jie Zhou, Yang Yang, Yi-Ling Wang, Yue Zhao, Wen-Jing Ye, Si-Yao Deng, Jin-Y Yi Lang, Shun Lu
Abstract
Key Words: Inflammatory bowel disease; Apoptosis; Enhancer of zeste homolog 2 ; JAK2 ;Permeability; Inflammatory bowel disease therapy
INTRODUCTION
Inflammatory bowel disease (IBD) is a worldwide health problem with an increasing incidence. IBD, including Crohn’s disease and ulcerative colitis and IBD unclassified,is described as relapsing, chronic gastrointestinal inflammation[1 ,2 ]. IBD patients experience low life quality and a higher risk of colorectal cancer[3 ]. The critical feature of IBD is the unsolved inflammation of the intestinal field caused by a breakdown to shift from a pro-inflammatory situation to an anti-inflammatory situation[4 ]. Various immune cell populations, such as eosinophils, neutrophils, dendritic cells, and macrophages, are present in the intestinal mucosa, participating in inflammation during IBD[5 ,6 ]. These cells participate in inflammation by secreting chemokines,antimicrobial agents, and pro-inflammatory cytokines, such as tumor necrosis factor(TNF)-α, interleukin (IL)-6 [3 ], and IL-1 β[7 ]. Therefore, the understanding of the molecular mechanism underlying the inflammatory regulation during IBD progression is urgently needed.
The fundamental function of epigenetic systems in determining T cell lineage fate decisions has been completely defined[8 ,9 ]. Nevertheless, the effect of the histone methyltransferase enhancer of zeste homolog 2 (EZH2 ) has been recently identified in the process[10 ,11 ]. EZH2 serves as a methylation of histone H3 K27 to H3 K27 me3 [12 ].It has been found that targeting EZH2 is able to alleviate intestinal inflammation in IBD[11 ]. Moreover, janus kinase 2 (JK2 )/ signal transducer and activator of transcription 3 (STAT3 ) signaling has been identified to alleviate inflammation response during IBD[13 ]. However, the correlation of EZH2 with JAK2 /STAT3 signaling in the IBD development remains unclear. In this study, we were interested in the molecular mechanism of EZH2 -mediated IBD progression.
MATERIALS AND METHODS
Cell culture
The NCM460 and fetal human colon (FHC) cell lines were maintained in the lab and were incubated at 37 °C with 5 % CO2 in Dulbecco’s Modified Eagle Medium (GE,United States) containing fetal bovine serum (15 %; GE Healthcare, Chicago, IL, United States), streptomycin (0 .1 mg/mL), and penicillin (100 units/mL). The lentiviral plasmids carrying EZH2 short hairpin ribonucleic acid (shRNA) and the corresponding control shRNA were obtained (GenScript, Nanjing, China). The transfection in the cells was performed by Liposome 3000 (Invitrogen, Carlsbad, CA, United States). The EZH2 inhibitor GSK343 (Sigma, St. Louis, MO, United States) and JAK2 inhibitor TG101348 (Selleck, Houston, TX, United States) were used at the dose of 5 μmol/L. The TNF-α (Sigma) was used at the dose of 50 ng/mL.
Clinical IBD samples
The clinical IBD samples (n= 50 ) and non-IBD control cases (n = 50 ) were collected from The Third People's Hospital of Chengdu. The diagnosis of IBD was consistent with the standard combination of radiologic, histological, endoscopic, and clinical criteria. The application of the samples was under the approval of the patients and proved by the Ethics Committee of Sichuan Cancer Hospital & Institute. The patients provided their written informed consent to participate in this study.
IBD mouse model
C57 BL/6 mice (female, 6 –8 wk) were obtained from the Chinese Academy of Medical Sciences (Beijing, China). The mice were randomly divided into three groups: Water group; dextran sodium sulfate (DSS) group; DSS + shEZH2 group. The mice in DSS and DSS + shEZH2 groups were constructed by adding DSS (2 .5 %, MP Biomedicals,Santa Ana, CA, United States) to drinking water for 7 d, followed by normal drinking water for the remaining days. The mice in DSS + shEZH2 groups were intraperitoneally injected with lentiviral plasmids carrying EZH2 shRNA (GenScript). The mice received DSS (3 %) to induce intestinal inflammation, and the natural death time of mice in the indicated groups within 15 d was recorded. The body weight was recorded at the indicated time. The colon length was measured and calculated using a ruler. The tissues were treated with 4 % paraformaldehyde, and the decalcification was conducted in 5 % nitric acid, followed by cutting into 5 μm sections. A set of alcohols was applied for the dehydration of the samples. Afterward, hematoxylin and eosin staining was performed in the slice samples of femoral head and then observed by microscope (BX-42 ; Olympus, Tokyo, Japan). The histologic score used to quantify the effect of EZH2 depletion in the mouse model by the pathologist was according to the criteria: Crypt architecture (normal, 0 –severe crypt distortion with loss of entire crypts,3 ), degree of inflammatory cell infiltration (normal, 0 –dense inflammatory infiltrate, 3 ),muscle thickening (base of crypt sits on the muscularis mucosae, 0 –marked muscle thickening, 3 ), crypt abscess (absent, 0 –present, 1 ), and goblet cell depletion (absent,0 –present, 1 )[1 ,2 ]. The levels of cytokines were measured by enzyme-linked immunosorbent assays (Sigma). Animal care was authorized by the Animal Ethics Committee. All experimental procedures with mice were performed in accordance and compliance with the regulations of the Laboratory Animal Welfare and Ethics Committee of Sichuan Cancer Hospital & Institute.
Analysis of cell apoptosis
About 2 × 105 NCM460 and FHC cells were plated on 6 -well dishes. Cell apoptosis was assessed by employing the Annexin V-FITC Apoptosis Detection Kit (Sigma) using the manufacture’s instruction. Shortly, about 2 × 105 collected and washed cells collected by binding buffer and were dyed at 25 °C, followed by the flow cytometry analysis.
Quantitative reverse transcription-polymerase chain reaction
Total RNAs were extracted using TRIZOL (Invitrogen). The first-strand complementary (c)DNA was manufactured as the manufacturer's instruction (TaKaRa, Kyoto,Japan). The quantitative reverse transcription-polymerase chain reaction was carried out by applying SYBR-Green (TaKaRa). The primer sequences are as follows: EZH2 forward: 5 ′-AATCAGAGTACATGCGACTGAGA-3 ′, reverse: 5 ′-GCTGTATCCTTCGCTGTTTCC; JAK2 forward: 5 ′-GCCTTCTTTCAGAGCCATCAT-3 ′, reverse:5 ′-GTGTAGGATCCCGGTCTTCAA-3 ′; Glyceraldehyde-3 -phosphate dehydrogenase forward: 5 ′-AACGGATTTGGTCGTATTGGG-3 ′, reverse: 5 ′-CCTGGAAGATGGTGATGGGAT-3 ′.
Western blot analysis
Total proteins were extracted from the cells using radioimmunoprecipitation assay buffer (Cell Signaling Technology, Danvers, MA, United States) and quantified using the BCA Protein Quantification Kit (Abbkine, Wuhan, China). The proteins at same concentration were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to poly(vinylidene fluoride) membranes (Millipore,Burlington, MA, United States), followed by the incubation with 5 % milk and with the primary antibodies at 4 °C overnight. The corresponding secondary antibodies (Boster,Wuhan, China) were used for incubating the membranes 1 h at room temperature,followed visualization by using chemiluminescence detection kit (Beyotime, Beijing,China). The primary antibodies applied in this study comprised EZH2 (Abcam,Cambridge, United Kingdom), Zona occludens 1 (ZO-1 ) (Abcam), claudin-5 (Abcam),occludin (Abcam), H3 K27 me3 (Abcam), JAK2 (Abcam), STAT3 (Abcam), p-JAK2 (Abcam), p-STAT3 (Abcam), EZH2 (Abcam), caspase3 , cleaved-caspase3 (Abcam), and β-actin (Abcam).
Chromatin immunoprecipitation analysis
Chromatin immunoprecipitation (ChIP) was performed using a SimpleChIP Enzymatic ChIP Kit (Cell Signaling Technology) according to the manufacturer's instruction. Chromatin prepared from the cells in a 15 cm dish was used to determine total DNA input and was incubated overnight with specific antibodies or normal rabbit immunoglobulin G. Then, the binding DNA was analyzed by quantitativepolymerase chain reaction assays; the primer sequences were shown as above.
Transepithelial electrical resistance measurements
Transepithelial electrical resistance (TEER) measurement was used to analyze the barrier function of the intestine. About 2 × 105 cells/mL cells were layered on collagencovered polycarbonate penetrable support supplements (Corning, Corning, NY,United States). The medium was replaced every 2 d. The production of the cells polarized monolayer at 3 wk was settled by monitoring and morphology of TEER by employing Millicell-ERS2 (Merck-Millipore). Delta-toxin was used to polarized monolayer cells at 37 °C.
Statistical analysis
The data were normally distributed. Data were expressed as mean ± SD, and the statistical analysis was conducted using GraphPad prism 7 (San Diego, CA, United States). The unpaired Student’st-test was used to compare two groups, and the oneway analysis of variance was used to compare multiple groups.P< 0 .05 was considered as statistically significant.
RESULTS
The depletion of EZH2 inhibits DSS-induced colitis in vivo
Firstly, we identified that the expression of EZH2 was enhanced in clinical IBD samples (n= 50 ) relative to healthy controls (n = 50 ) (Figure 1 A). Meanwhile, we found that DSS enhanced EZH2 expression in mice, while the depletion of EZH2 by shRNA repressed the enhancement (Figure 1 B and C). We then measured the effect of EZH2 on the DSS-induced colitisin vivo. We observed that the DSS treatment significantly resulted in a weight loss of the mice, which was attenuated by EZH2 depletion(Figure 1 D). The colon length was reduced in the DSS-treated mice and was rescued by the EZH2 depletion in the system (Figure 1 E and F). Meanwhile, the DSS treatment caused a decreased histological score in the mice, which was rescued by EZH2 depletion (Figure 1 G). Moreover, the inflammatory cytokines, such as IL-6 , IL-1 β, and TNF-α, were enhanced in the DSS-treated mice, in which the depletion of EZH2 could reverse this effect (Figure 1 H-J).


Figure 1 The depletion of enhancer of zeste homolog 2 inhibits dextran sodium sulfate-induced colitis in vivo. A: The expression of enhancer of zeste homolog 2 (EZH2 ) was measured by quantitative real-time polymerase chain reaction in clinical inflammatory bowel disease (IBD) samples (n = 50 ) and healthy controls (n = 50 ); B-J: The IBD mouse model was conducted by adding dextran sodium sulfate (DSS). The mice were intraperitoneally injected with lentiviral plasmids carrying EZH2 shRNA; B: The expression of EZH2 was detected by quantitative real-time polymerase chain reaction in the mice; C: The protein levels of EZH2 were analyzed by Western blot in the mice; D: The body weight in the mice; E and F: Colon length of the mice; G: representative hematoxylin and eosin staining of distal colon sections; H-J: Colonic inflammatory cytokines in the mice. n = 10 , mean ± SD. aP < 0 .05 ; bP < 0 .01 ; cP < 0 .001 . TNF-α: Tumor necrosis factorα; IL: Interleukin.
EZH2 depletion attenuates apoptosis of colonic epithelial cells
Then, the NCM460 and FHC colonic epithelial cells were treated with TNF-α or cotreated with TNF-α and EZH2 shRNA. We found that the expression of cleavedcaspase-3 was enhanced in the TNF-α-treated NCM460 and FHC cells, and the depletion of EZH2 was able to inhibit the enhancement in the cells (Figure 2 A and B).Moreover, the TNF-α treatment induced the apoptosis of NCM460 and FHC cells, in which EZH2 knockdown could reverse this effect in the cells (Figure 2 C and D).
The depletion of EZH2 attenuates permeability of colonic epithelial cells
Next, we further identified that the TEER was reduced by TNF-α treatment in the NCM460 and FHC cells, which was rescued by the depletion of EZH2 in the cells(Figure 3 A and B). Consistently, the expression of permeability markers, including ZO-1 , claudin-5 , and occludin, was inhibited in the TNF-α-treated NCM460 and FHC cells, but the EZH2 depletion could rescue this phenotype in the cells (Figure 3 C and D).
EZH2 inactivates JAK2 /STAT3 signaling in colonic epithelial cells
Significantly, Western blot analysis showed that the depletion of EZH2 enhanced the expression and the phosphorylation of JAK2 and STAT3 in the NCM460 and FHC cells(Figure 4 A and B). Meanwhile, the expression and the phosphorylation of JAK2 and STAT3 EZH2 inhibitor were induced by the EZH2 inhibitor GSK343 in the cells(Figure 4 C and D).

Figure 2 Enhancer of zeste homolog 2 depletion attenuates apoptosis of colonic epithelial cells. The NCM460 and fetal human colon (FHC) cells were treated with tumor necrosis factor-α (TNF-α), or co-treated with TNF-α and enhancer of zeste homolog 2 (EZH2 ) short hairpin RNA. A and B: Western blot analysis of the caspase3 and cleaved-caspase3 expression; C and D: Flow cytometry analysis of the cell apoptosis. n = 3 , mean ± SD, bP < 0 .01 .
EZH2 inactivates JAK2 expression by regulating histone H3 K27 me3
Furthermore, we observed that the H3 K27 me3 was reduced, along with the enhanced expression and the phosphorylation of JAK2 and STAT3 , in the EZH2 -knockdown NCM460 and FHC cells (Figure 5 A and B). In addition, the depletion of EZH2 reduced the levels of H3 K27 me3 on the promoter of JAK2 in the cells (Figure 5 C and D).Consistently, the mRNA expression of JAK2 was up-regulated by EZH2 depletion in the NCM460 and FHC cells (Figure 5 E and F).
EZH2 promotes apoptosis and permeability by inactivating JAK2 /STAT signaling in colonic epithelial cells
Next, we identified that EZH2 inhibitor GSK343 attenuated TNF-α-induced apoptosis of the NCM460 and FHC cells, and the JAK2 inhibitor TG101348 was able to rescue the phenotype in the cells (Figure 6 A and B). Moreover, GSK343 rescued TNF-α-inhibited ZO-1 , claudin-5 , and occludin expression in the NCM460 and FHC cells, in which the co-treatment with TG101348 could reverse this effect in the system (Figure 6 C and D).

Figure 3 The depletion of enhancer of zeste homolog 2 attenuates permeability of colonic epithelial cells. The NCM460 and fetal human colon(FHC) cells were treated with tumor necrosis factor-α (TNF-α), or co-treated with TNF-α and enhancer of zeste homolog 2 (EZH2 ) short hairpin RNA. A and B:Transepithelial electrical resistance (TEER) measurement of the transepithelial electrical resistance levels; C and D: Western blot analysis of the Zona occludens 1 (ZO-1 ), claudin-5 , and occludin expression. n = 3 , mean ± SD, bP < 0 .01 .
DISCUSSION
IBD is a prevalent inflammatory gastrointestinal disease with high incidence. As a critical epigenetic regulator, EZH2 has been identified to contribute to IBD progression, but the mechanism remains unclear. In this study, we found that EZH2 promoted apoptosis and inflammatory response by inactivating JAK2 /STAT signaling in IBD.
Several previous investigations have shown the function of EZH2 in the modulation of IBD. It has been reported that EZH2 is involved in the pathobiological mechanism of IBD progression[14 ]. Inhibiting EZH2 attenuates intestinal inflammation of IBD development[11 ]. EZH2 is an epigenetic regulator in colitis by inhibiting TNF-αregulated apoptosis and inflammation in IBD[15 ]. EZH2 modulates intestinal necroptosis and inflammation in intestinal epithelial cells by c-jun N-terminal kinase signaling[16 ]. Targeting EZH2 enhances cAMP response element-binding protein to inhibit the ulcerative colitis progression[17 ]. It has been well-recognized that EZH2 presents a critical biological significance of in autoimmune disorders and cancer, and thereby targeting EZH2 is a promising therapeutic strategy in these diseases[18 ,19 ]. In this study, we demonstrated that the depletion of EZH2 inhibited DSS-induced colitisin vivo.EZH2 depletion attenuated apoptosis and permeability of colonic epithelial cells. Our data identified a critical role of EZH2 in the IBD progression, providing informative evidence of the function of EZH2 in modulating IBD development.
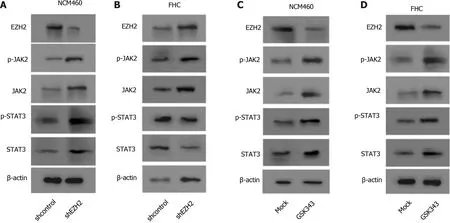
Figure 4 Enhancer of zeste homolog 2 inactivates janus kinase 2 / signal transducer and activator of transcription signaling in colonic epithelial cells. A and B: The NCM460 and fetal human colon (FHC) cells were treated with control shRNA or enhancer of zeste homolog 2 (EZH2 ) short hairpin RNA. Western blot analysis of the Janus kinase 2 (JAK2 ), Signal transducer and activator of transcription 3 (STAT3 ), and EZH2 expression and JAK2 and STAT3 phosphorylation; C and D: The NCM460 and FHC cells were treated with GSK343 (5 μM). Western blot analysis of the JAK2 , STAT3 , and EZH2 expression and JAK2 and STAT3 phosphorylation. n = 3 .

Figure 5 Enhancer of zeste homolog 2 inactivates janus kinase 2 expression by regulating histone H3 K27 me3 . A-E: The NCM460 and fetal human colon (FHC) cells were treated with control short hairpin RNA or enhancer of zeste homolog 2 (EZH2 ) short hairpin RNA; A and B: Western blot analysis of the H3 K27 me3 , Janus kinase 2 (JAK2 ), signal transducer and activator of transcription 3 (STAT3 ), and EZH2 expression and JAK2 and STAT3 phosphorylation; C and D: Chromatin immunoprecipitation (ChIP) assays of the JAK2 promoter using H3 K27 me3 antibody; E and F: The quantitative real-time polymerase chain reaction assays of JAK2 messenger RNA expression. n = 3 , mean ± SD, bP < 0 .01 .

Figure 6 Enhancer of zeste homolog 2 promotes apoptosis and permeability by inactivating janus kinase 2 / signal transducer and activator of transcription signaling in colonic epithelial cells. The tumor necrosis factor-α (TNF-α)-treated NCM460 and fetal human colon (FHC) cells were treated with GSK343 (5 μM) or co-treated with GSK343 (5 μM) and TG101348 (5 μM). A and B: Flow cytometry analysis of the cell apoptosis; C and D: Western blot analysis of the Zona occludens 1 (ZO-1 ), claudin-5 , and occludin expression. n = 3 , mean ± SD. bP < 0 .01 .
As essential cellular signaling in various pathological conditions, JAK2 /STAT3 signaling plays critical role in IBD regulation. It has been found that V617 F is able to up-regulate JAK2 expression in patients with IBD[20 ]. Sphk1 enhances ulcerative colitis by modulating JAK2 /STAT3 signaling[21 ]. Renin-angiotensin system contributes to colonic inflammation by inducing helper T 17 activation through JAK2 /STAT signaling[22 ]. A20 inhibits IBDviarepressing nuclear factor-kappa B and STAT3 in mice[23 ]. Our mechanical studies showed that EZH2 inactivated JAK2 expression by regulating histone H3 K27 me3 . EZH2 promoted apoptosis and permeability by inactivating JAK2 /STAT signaling in colonic epithelial cells. These data discover an unreported association of EZH2 with JAK2 /STAT signaling,elucidating a new mechanism involving EZH2 and JAK2 /STAT in IBD pathogenesis.
CONCLUSION
In conclusion, we discovered that EZH2 contributed to apoptosis and inflammatory response by inactivating JAK2 /STAT signaling in IBD. EZH2 is a promising target for treatment of IBD.
ARTICLE HIGHLIGHTS

杂志排行

World Journal of Gastroenterology的其它文章
- Fecal microbiota transplantation for irritable bowel syndrome: An intervention for the 21 st century
- Hepatocellular carcinoma in viral and autoimmune liver diseases: Role of CD4 + CD25 + Foxp3 + regulatory T cells in the immune microenvironment
- Application of artificial intelligence-driven endoscopic screening and diagnosis of gastric cancer
- Mucosal lesions of the upper gastrointestinal tract in patients with ulcerative colitis: A review
- Preservation of superior rectal artery in laparoscopically assisted subtotal colectomy with ileorectal anastomosis for slow transit constipation
- Early serum albumin changes in patients with ulcerative colitis treated with tacrolimus will predict clinical outcome