染料中富马酸二甲酯的检测
2021-06-07赵广明刘秋红任祖光
赵广明,吕 艳,刘秋红,任祖光
(浙江龙盛染料化工有限公司,浙江绍兴 312300)
富马酸二甲酯(dimethyl fumarate,DMFu)又称别马来酸二甲酯(allomaleicacid-dimethylester),俗称防霉保鲜剂霉克星1号;常温下为白色结晶或者结晶粉末,溶于乙酸乙酯、氯仿、丙酮和醇类,微溶于乙醚、水。富马酸二甲酯具有高效、广谱抗菌的特点,对霉菌有特殊的抑菌效果,兼有杀虫活性,还具有触杀和熏蒸作用,曾广泛应用于食品、饮料、饲料、中药材、化妆品、鱼、肉、蔬菜、水果等的防霉、防腐、防虫、保鲜[1]。
随着人们对富马酸二甲酯的深入研究,实验发现富马酸二甲酯具有一定毒性且会慢性积累,进入人体会对器官产生腐蚀性损害,尤其是对儿童的成长发育造成严重危害。2009年1月29日,欧盟成员国通过了“保证含有富马酸二甲酯的消费品不会投放欧洲市场”的决议草案,该决议于2009年5月1日正式生效。草案明确规定,如果消费品或其部件中富马酸二甲酯的质量分数超过了0.1 mg/kg,或者产品本身已声明了其富马酸二甲酯的质量分数,就将被认定为“含有富马酸二甲酯”的产品,禁止进入欧盟市场流通和销售。2009年2月,中国禁止将富马酸二甲酯作为食品添加剂使用,并且在皮革、鞋类和纺织品及食品上制定了相关检测标准。在不同领域采用的检测标准不同,王云玉等[2]采用顶空-GC-MS法对皮革及其制品中的富马酸二甲酯含量进行检测;孙忠松等[3]等用乙腈提取纺织品及皮革中的富马酸二甲酯;国家建立了纺织品中富马酸二甲酯的国家标准GB/T 28190—2011《纺织品富马酸二甲酯的测定》。作为纺织印染的上游行业——染料行业,富马酸二甲酯检测的相关标准亟待建立。本项目通过对染料产品中富马酸二甲酯提取方法和色谱、质谱条件的改进,建立了染料中富马酸二甲酯的气相色谱-质谱联用检测方法。
1 实验
1.1 试剂和仪器
试剂:丙酮、N,N-二甲基甲酰胺、乙酸乙酯、乙醇、乙腈(分析纯),标准品富马酸二甲酯,标准品富马酸二甲酯D2。
仪器:Phenom ProX电镜能谱一体机(SEM电镜扫描仪,荷兰飞纳),7890B-5977A安捷伦气相色谱-质谱联用仪(带自动进样器G4567A,美国安捷伦),Phenomenex ZB-35MS色谱柱(美国),微量注射器、超声波振荡器(宁波新芝生物科技有限公司),台式低速离心机(4 000 r/min,上海医疗器械有限公司)。
1.2 萃取步骤
使用有机溶剂萃取染料产品中的富马酸二甲酯,用气相色谱仪分离萃取液中的组分,质谱仪定性,用富马酸二甲酯D2作为内标,再用峰面积内标法定量测定染料中的富马酸二甲酯质量分数。
1.2.1 质谱操作条件
仪器操作条件如表1所示。可根据仪器设备不同,选择最佳分析条件。
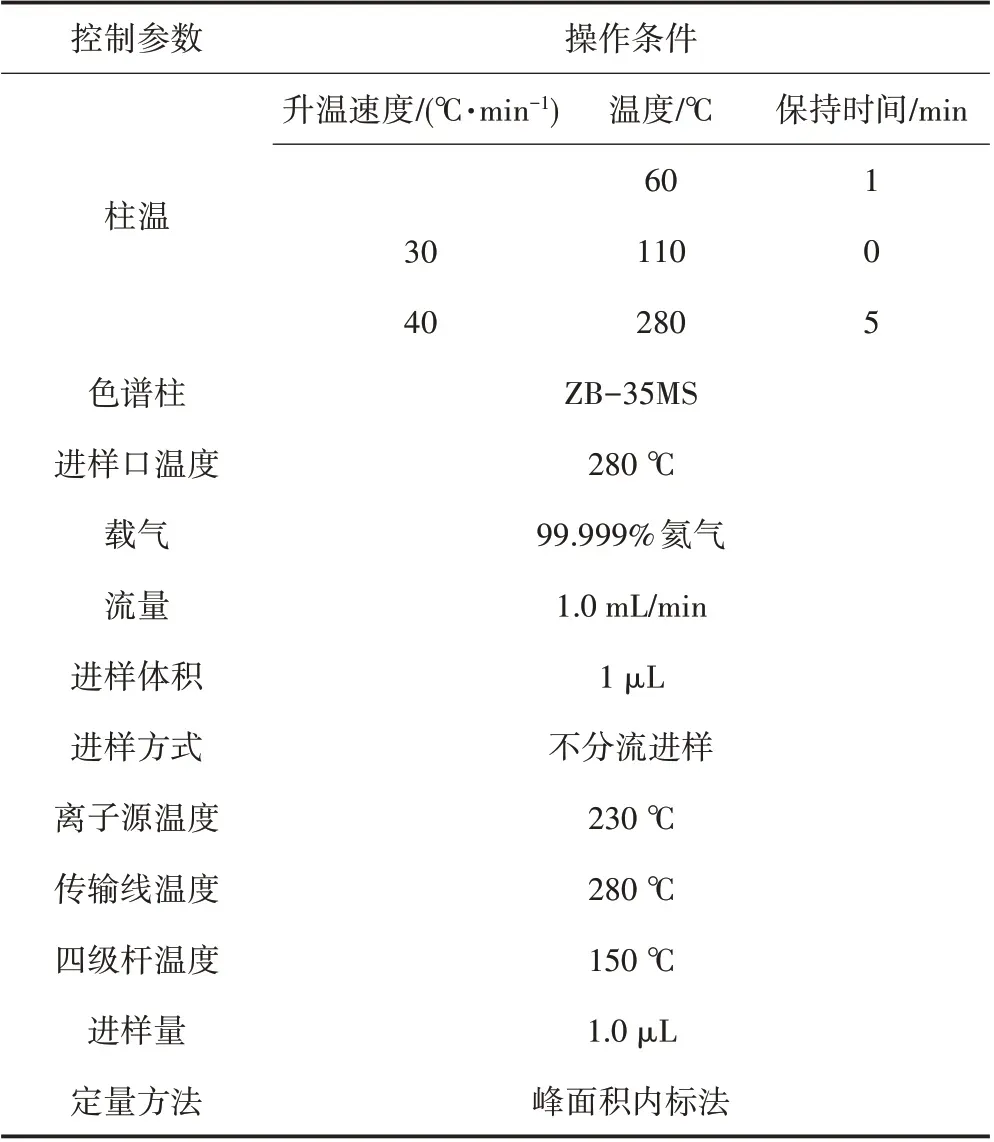
表1 仪器操作条件
1.2.2 标准样品的配制
准确称取标样富马酸二甲酯、内标物富马酸二甲酯D2各0.01 g(准确至0.000 1 g),用丙酮溶解后,分别定容至100 mL容量瓶中。此溶液为标准储备溶液,质量浓度约为100 mg/L,-10~-5℃避光保存,保存期为半年。
分别吸取相同适量的两种标准储备溶液至50 mL容量瓶中,用丙酮定容至刻度,作为混合标准工作溶液,-10~-5℃避光保存,保存期为3个月。
1.2.3 样品前处理
准确称取样品0.5 g(精确至0.000 1 g),置于40 mL样品瓶中,加入10 mL丙酮内标液(内标物质量分数为0.1 mg/kg),于超声波发生器中带温超声30 min(温度为50℃)。冷却至室温后,取约5 mL样品溶液于离心管中,用离心机离心5 min后,取上层清液,上机检测。
1.3 测试
根据染料样品中被测物的质量分数,选取质量分数相近的标准工作溶液进行测定。如果样品中富马酸二甲酯的质量分数太高,则将前处理好的样品稀释至常用标准溶液的质量分数再进行测定。按上述质谱仪分析条件,用自动进样器分别取1.0μL试样溶液和标准工作溶液进行测定,待出峰完毕后,用质谱工作站进行结果处理,通过比较试样与标样色谱峰的保留时间和质谱选择特征离子进行定性,用峰面积内标法进行定量。
校正因子计算式:
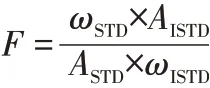
式中,F为校正因子;ωSTD为标样富马酸二甲酯的质量分数,mg/kg;ωISTD为内标物富马酸二甲酯D2的质量分数,mg/kg;ASTD为标样富马酸二甲酯的峰面积;AISTD为内标物富马酸二甲酯D2的峰面积。
富马酸二甲酯的质量分数按下式计算:
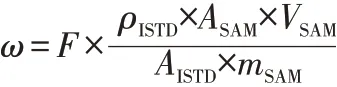
式中,ω为试样中富马酸二甲酯的质量分数,mg/kg;F为校正因子;ρISTD为内标物富马酸二甲酯D2的质量浓度,mg/L;mSAM为试样的质量,g;AISTD为内标物富马酸二甲酯D2的峰面积;ASAM为试样中富马酸二甲酯的峰面积;VSAM为试样萃取液的体积,mL。试样中富马酸二甲酯的质量分数结果保留小数点后两位。样品中富马酸二甲酯的质谱图见图1。
图1 样品中富马酸二甲酯的色谱图
2 结果与讨论
2.1 样品的前处理方法优化
2.1.1 溶剂的选择
实验选择不同溶剂(丙酮、N,N-二甲基甲酰胺、乙酸乙酯、乙腈、乙醇)直接进行萃取。0.5 g染料样品在不同溶剂中的溶解情况如表2所示。

表2 染料样品在不同溶剂中的溶解情况
由表2可知,由于分散染料难溶于水和常规溶剂,一般对溶剂的选择不能只考虑富马酸二甲酯的溶解性,还需要考虑分散染料以及分散剂是否溶解。分散染料普遍采用喷干技术制备[4],通过电镜扫描观察染料结构,染料粒径为50~200μm(如图2所示),溶剂如果不能将染料溶解,染料颗粒仍旧存在,因为部分富马酸二甲酯被包含在染料颗粒中,无法完全溶解,会造成测试结果偏小甚至无法检测。
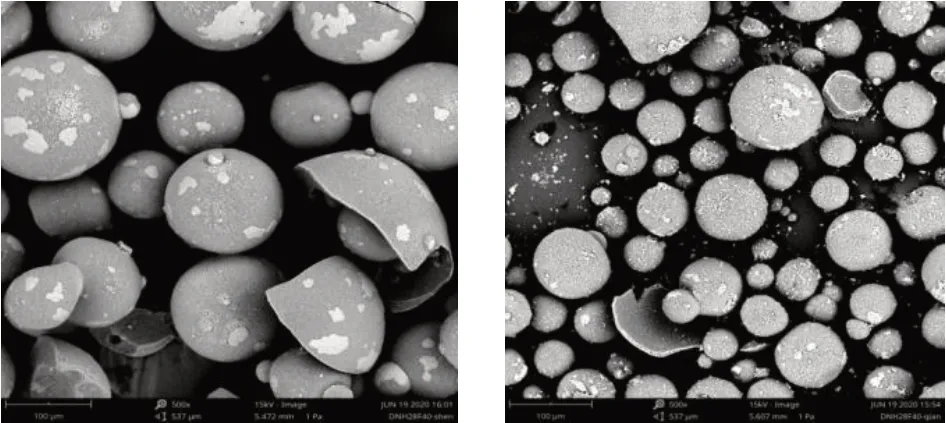
图2 染料放大500倍
富马酸二甲酯在N,N-二甲基甲酰胺中溶解度最低,在丙酮中溶解度最高,选取溶解度最高的丙酮和溶解度最低的N,N-二甲基甲酰胺以及常用的乙酸乙酯、乙醇作为溶剂进行实验,结果如表3所示。由表3可以看出,富马酸二甲酯在丙酮中测试结果最高,效果最好。因此,采用丙酮作为溶剂对样品进行前处理。
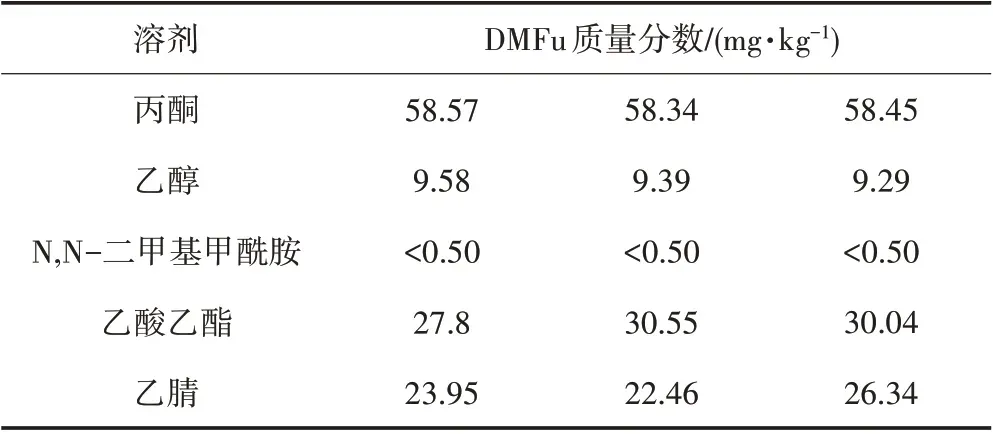
表3 不同溶剂对测试结果的影响
2.1.2 超声温度和时间
由表4可知,超声时间越长、温度越高,检测出染料中富马酸二甲酯的质量分数越高,由于丙酮的沸点是56.5℃,因此选择超声温度50℃、时间30 min。
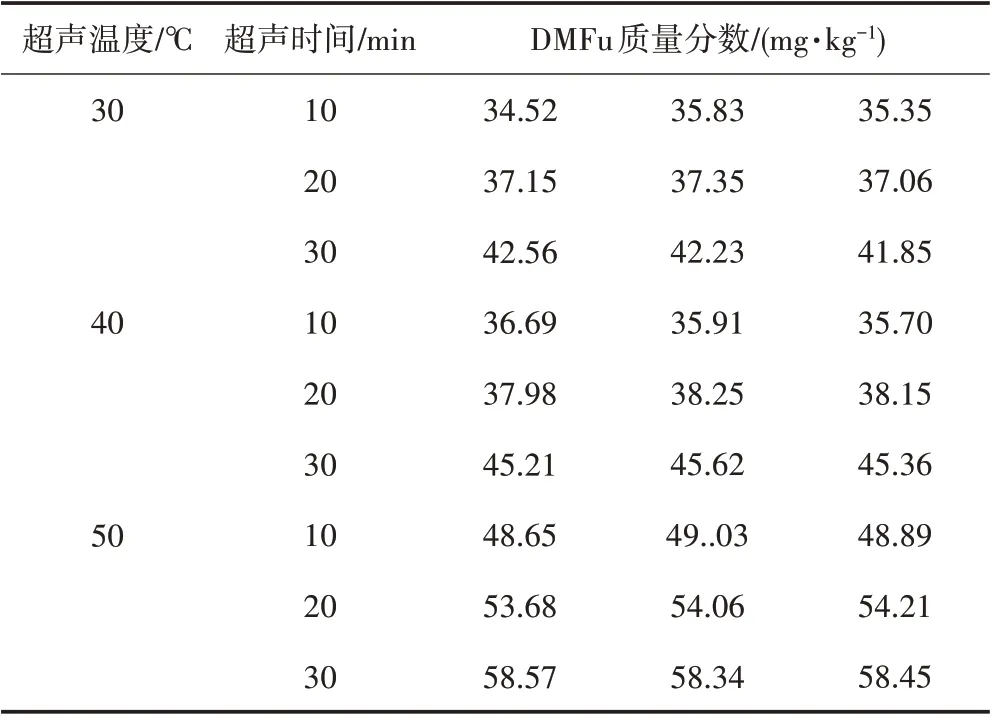
表4 超声温度和时间对测试结果的影响
2.2 气相色谱分离条件的优化
由表5可以看出,使用ZB-35MS(30 m×0.25 mm×0.25μm)色谱柱分离前处理后的样品,杂质与富马酸二甲酯分离效果好。高温区300℃保持5 min可使样品中沸点较高的组分流出色谱系统,避免质谱柱污染。

表5 色谱柱分离效果对比
2.3 富马酸二甲酯的定性
测试样品与标准工作溶液的选择离子总流图中,内标物富马酸二甲酯D2出峰时间与富马酸二甲酯出峰时间一样,在测试样品与标准工作液溶相同的保留时间出现的色谱峰分别为提取富马酸二甲酯和内标物富马酸二甲酯D2的特征离子碎片,根据其丰度比进行确证。
气相色谱-质谱联用仪选择离子色谱总流图和质谱图如图3~8所示。
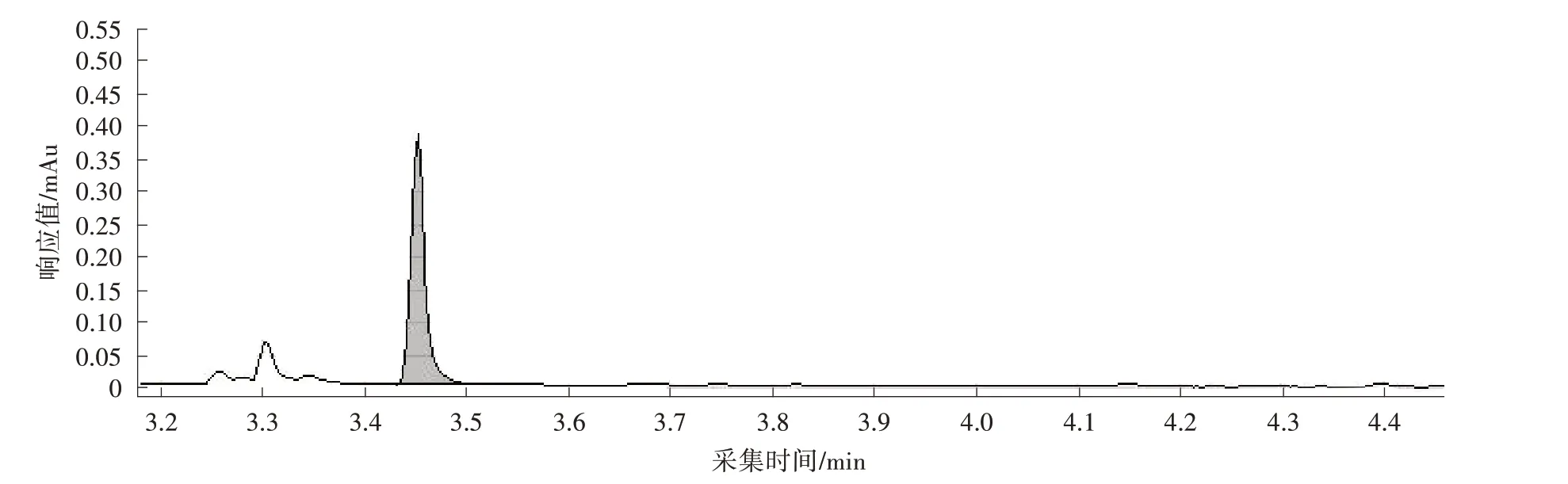
图3 富马酸二甲酯色谱图
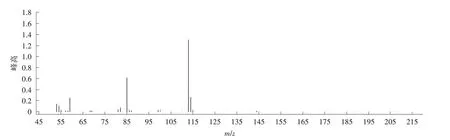
图4 富马酸二甲酯质谱图
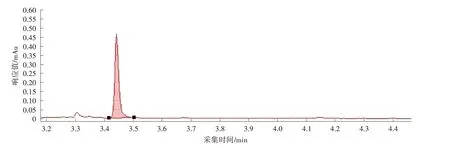
图5 富马酸二甲酯D2色谱图
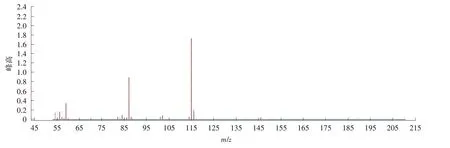
图6 富马酸二甲酯D2质谱图
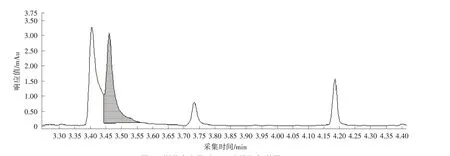
图7 样品中富马酸二甲酯提取色谱图

图8 样品中富马酸二甲酯D2提取色谱图
2.4 仪器的稳定性和方法的精密度
将富马酸二甲酯标准储备溶液逐级稀释,然后配制成不同质量分数的标准溶液,最后再以富马酸二甲酯的质量分数为横坐标,以富马酸二甲酯的峰面积为纵坐标,对富马酸二甲酯绘制标准曲线,结果如图9所示。
由图9可以看出,富马酸二甲酯的质量分数在0.05~2.00 mg/kg内和峰面积具有良好的线性关系,相关系数接近1,能够满足检验工作要求。
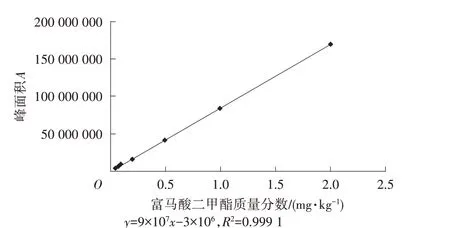
图9 富马酸二甲酯的标准曲线
在不含富马酸二甲酯的染料中加入一定量的标准工作溶液,在本方法的前处理操作条件下重复进样10次,记录标样峰面积,结果见表6。依据JJF 1164—2018气相色谱-质谱联用仪校准规范,指标为小于等于10%。由表6可以看出,在本方法确定的条件下,分析仪器具有良好的稳定性,方法具有良好的精密度。
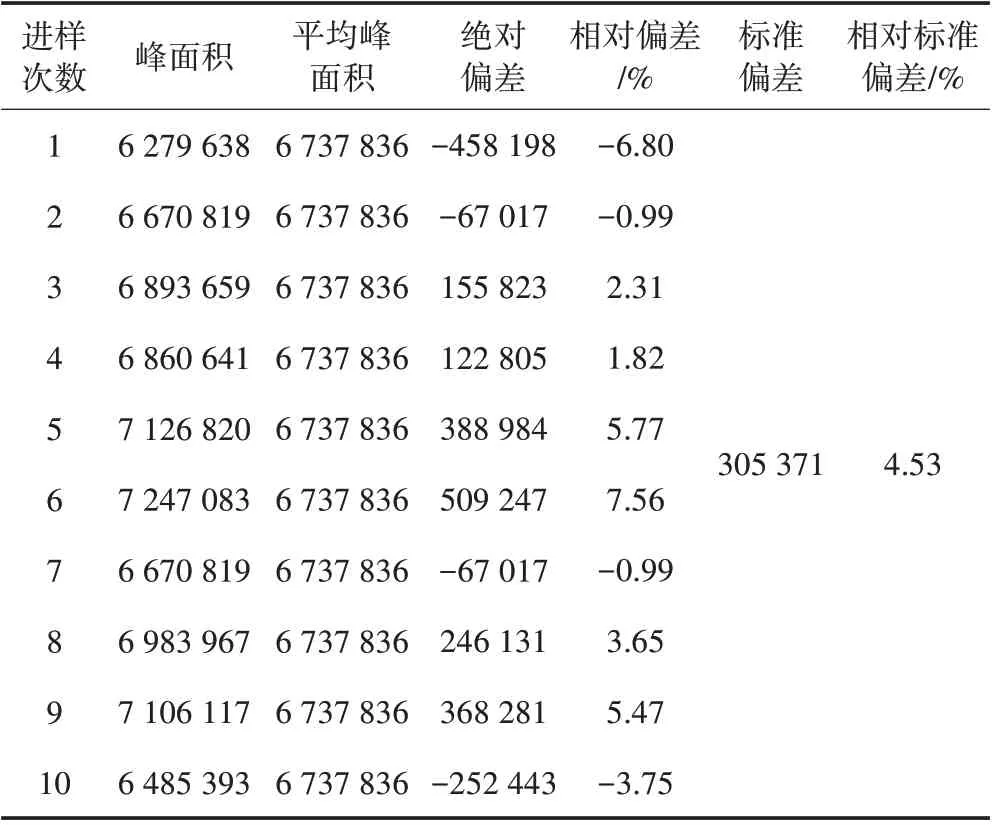
表6 仪器的稳定性及方法的精密度
2.5 方法回收率
采用标准加入法,将1 mL混合标液加入到0.5 g不含富马酸二甲酯的染料中,按照本方法操作,测得富马酸二甲酯质量分数的回收率为70%~130%。
2.6 最小定量值
在本方法的线性范围内,以10倍信噪比峰高的目标化合物作为最小定量值。本方法根据多次实验数据可得:染料中富马酸二甲酯的最小定量限值为0.5 mg/kg,低于此限值作为未检出处理。
3 结论
本方法采用丙酮作为溶剂,以气相色谱-质谱联用仪器检测染料样品中富马酸二甲酯的质量分数,具有提取简单、准确、稳定,回收率和精密度高,干扰少等优点,适用于染料中富马酸二甲酯的检测。检测染料中富马酸二甲酯的质量分数具有现实意义,对其他类似化学品中富马酸二甲酯的测试具有参考价值。
