H2AX磷酸化抑制肺癌细胞发生上皮-间质转化的作用机制
2021-06-04乔婷婷葛淑静陆承荣段连宁
乔婷婷,葛淑静,罗 渊,陆承荣,段连宁
1.中国人民解放军空军特色医学中心临床医学实验室,北京 100142;2.中国人民解放军空军特色医学中心干部病房,北京 100142;3.中国人民解放军空军特色医学中心保健诊疗科,北京 100142
侵袭转移是临床肿瘤治疗失败的主要原因,导致90%以上实体瘤患者死亡。而癌细胞的上皮-间质转化(epithelial-to-mesenchymal transition,EMT)是肿瘤转移启动和发生的关键生物学过程[1-2]。EMT一旦启动,肿瘤细胞之间的黏附能力失去,使得肿瘤细胞获得迁移和侵袭能力,并向远端器官或组织转移。因此,抑制EMT无疑是治疗肿瘤转移的重要选择。EMT是一个复杂的多步骤程序性调控过程,其关键性调控机制有待于进一步详细阐明。EMT过程涉及多种基因调控包括Snail、Slug、Zeb1/2、vimentin、THBS1、VCAN、TGFB2、Twist等间质基因的上调以及上皮基因E-cadherin的下调[3-4]。根据报道,血管内皮生长因子(vascular endothelial growth factor,VEGF)/血管内皮生长因子受体(vascular endothelial growth factor receptor,VEGFR)信号转导通路直接参与肿瘤转移过 程[5-6]。肿瘤细胞分泌VEGF刺激血管内皮细胞膜上的VEGFR,诱导肿瘤组织血管生成,肿瘤细胞自分泌VEGF刺激其细胞膜上VEGFR,并激活其下游通路raf/MEK/MAPK进而刺激肿瘤细胞启动EMT和转移[7-8]。临床标本检测也显示,VEGFR家族(VEGFR-1、VEGFR-2和VEGFR-3)在肿瘤组织高表达,但其表达机制不清楚。
H2AX是组蛋白H2A家族的一个变体分子。大量早期研究表明,磷酸化H2AX(γH2AX)被认为参与基因组DNA分子损伤修复[9-10]。中国学者陆承荣等[1]于2006年首次发现H2AX具有调控凋亡的作用,基因敲除H2AX后细胞凋亡通路受阻,并详细揭示丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)家族c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)介导H2AX磷酸化进而调控凋亡的分子机制[11]。由于H2AX具有调控凋亡的关键作用,相关研究也在不断扩展,尤其与人类重大疾病肿瘤的关系也陆续被证明。目前已有大量报道包括本课题组发表的数据,均证明H2AX在肿瘤细胞凋亡时发挥调控作用[12-14]。最近,我们又发现了H2AX新的功能。当抑制小细胞肺癌细胞内H2AX时,则强烈刺激肺癌细胞发生EMT;结果明确提示,在肺癌细胞EMT过程中H2AX发挥调控作用,H2AX磷酸化抑制肺癌EMT,H2AX去磷酸化后则促进EMT。目前有关H2AX与EMT关系的研究少见报道,尤其H2AX在肺癌转移中的作用未见发表。本研究深入探讨H2AX磷酸化修饰表观遗传调控肺癌转移的新功能,从全新角度认识肺癌细胞EMT和转移的分子机制,为肺癌等其他肿瘤转移治疗提供新的靶向性和特异性更强的候选分子靶点。
1 材料和方法
1.1 细胞来源
人肺癌A549细胞由中国农业大学扈洪波馈赠;A549稳定株细胞包括表达野生型H2AX(H2AX-wild type,H2AX-wt)、表达Ser139磷酸化位点突变H2AX(H2AX-139m)、基因沉默稳定细胞株H2AX(H2AX-knockdown,H2AXkd)由本实验室前期制备[15];H2AX野生型(H2AX+/+)和H2AX基因敲除(H2AX-/-)小鼠胚胎成纤维细胞(mouse embryonic fibroblast,MEF)由美国国家癌症研究所Andre Nussenzweig提供[11]。
1.2 蛋白质印迹法(Western blot)检测
细胞总蛋白提取如前所述。蛋白样品通过十二烷基硫酸钠聚丙烯酰胺凝胶电泳(sodium dodecyl sulphate polyacrylamide gel electrophoresis,SDS-PAGE)分离,并转到PVDF膜(美国Millipore公司)上,然后把PVDF膜放入洗膜缓冲液(Tris-buffered saline Tween,TBST)配制的5%脱脂奶粉中,室温下封闭1 h。封闭后的PVDF膜与H2AX磷酸化抗体、E-cadherin抗体、vimentin抗体、H2AX抗体、β-actin抗体(美国Millipore公司)在4 ℃条件下温育过夜,然后与TBST(5%脱脂奶粉)配制的二抗(日本MBL公司)在室温下温育1 h。一抗浓度为1∶1 000,二抗浓度为1∶2 000。利用化学发光试剂SignalFire ECL(美国Cell Signaling Technology公司)检测蛋白。
1.3 mRNA提取及实时荧光定量聚合酶链反应(real-time fluorescence quantitative polymerase chain reaction,RTFQ-PCR)
总RNA使用TRIzol试剂(美国LifeTechnologies公司)提取。反转录使用FastQuant RT试剂盒(含gDNA)按说明书执行[天根生化科技(北京)有限公司]。E-cadherin mRNA、vimentin mRNA、Slug mRNA、β-cantenin mRNA及β-actin mRNA反转录按说明书执行。RTFQPCR使用SuperRealPreMix Plus试剂盒按厂家说明书执行[天根生化科技(北京)有限公司]。β-actin mRNA作为内对照。E-cadherin的有义链序列为5’-TGGAGGAATTCTTGCTTTGC-3,反义链序列为5’-CGCTCTCCTCCGAAGAAAC-3’;vimentin的有义链序列为5’-TGGTCTAACGGTTTCCCCTA-3’,反义链序列为5’-GACCTCGGAGCGAGAGTG-3’;Slug的有义链序列为5’-TGGTTGCTTCAAGGACACAT-3’,反义链序列为5’-GCAAATGCTCTGTTGCAGTG-3’;β-cantenin的有义链序列为5’-GCTTTCAGTTGAGCTGACCA-3’,反义链序列为5’-CAAGTCCAAGATCAGCAGTCTC-3’;β-actin的有义链序列为5’-CCAACCGCGAGAAGATGA-3’,反义链序列为5’-CCAGAGGCGTACAGGGATAG-3’。RTFQ-PCR反应条件:95℃变性10s,退60℃火20s,延伸72℃10s,循环40次。
1.4 siRNA和转染
VEGFR-2 siRNA和对照siRNA(CTR siRNA)以及转染所需配套试剂均购自美国Santa Cruz Biotechnology公司,具体操作步骤按照说明书进行。首先将含有siRNA的溶液A直接加入到转染稀释液中,轻轻混匀,在室温下温育15~ 45 min待用。然后用siRNA转染培养基清洗肺癌A549细胞1次,并加入siRNA转染试剂混合物(溶液A+溶液B),在37 ℃、CO2培养箱中培养细胞5~7 h。
1.5 EMT诱导和检测
肺癌A549稳定株细胞或经过siRNA转染的A549细胞血清饥饿24 h后,以5%血清刺激,诱导EMT,然后提取细胞总蛋白和总mRNA,检测EMT标志性分子E-cadherin和vimentin的蛋白(Western blot)和mRNA水平(RTFQ-PCR),肺癌细胞的迁移和侵袭能力使用transwell试剂(美国Corning公司)按说明书进行。
1.6 统计学处理
所有实验过程均进行3次独立重复实验,实验数据用表示(为3次独立重复实验的平均值,s为实验的标准差),所得数据应用SPSS 22.0进行分析,分析时采用t检验或ANOVA进行分析,P<0.05为差异有统计学意义。
2 结果
2.1 H2AX基因沉默刺激肺癌A549细胞发生EMT
为了揭示H2AX是否参与调控肺癌A549细胞的EMT过程,我们用5%胎牛血清刺激肺癌A549细胞H2AX-kd及其对照细胞稳定株(CTR),然后用transwell方法检测肺癌A549细胞的迁移和侵袭能力。结果显示,H2AX基因沉默下调H2AX后显著增强A549细胞的迁移(图1A)和侵袭(图1B)能力,说明H2AX抑制A549细胞EMT。为了进一步明确H2AX对肺癌细胞EMT的作用,我们提取稳定株细胞CTR和H2AX-kd的总蛋白和总RNA,检测EMT发生的标志性分子E-cadherin和vimentin的蛋白和mRNA水平。Western blot和RTFQ-PCR结果一致,均说明血清刺激诱导EMT后,H2AX-kd细胞内E-cadherin蛋白(图1C)和E-cadherin mRNA(图1D)水平下调,而vimentin蛋白(图1C)和vimentin mRNA(图1D)上调。在H2AX-kd细胞内H2AX蛋白抑制后,H2AX磷酸化也下调(图1C)。总之,以上实验结果明确说明,沉默H2AX基因可以刺激肺癌A549细胞发生EMT。
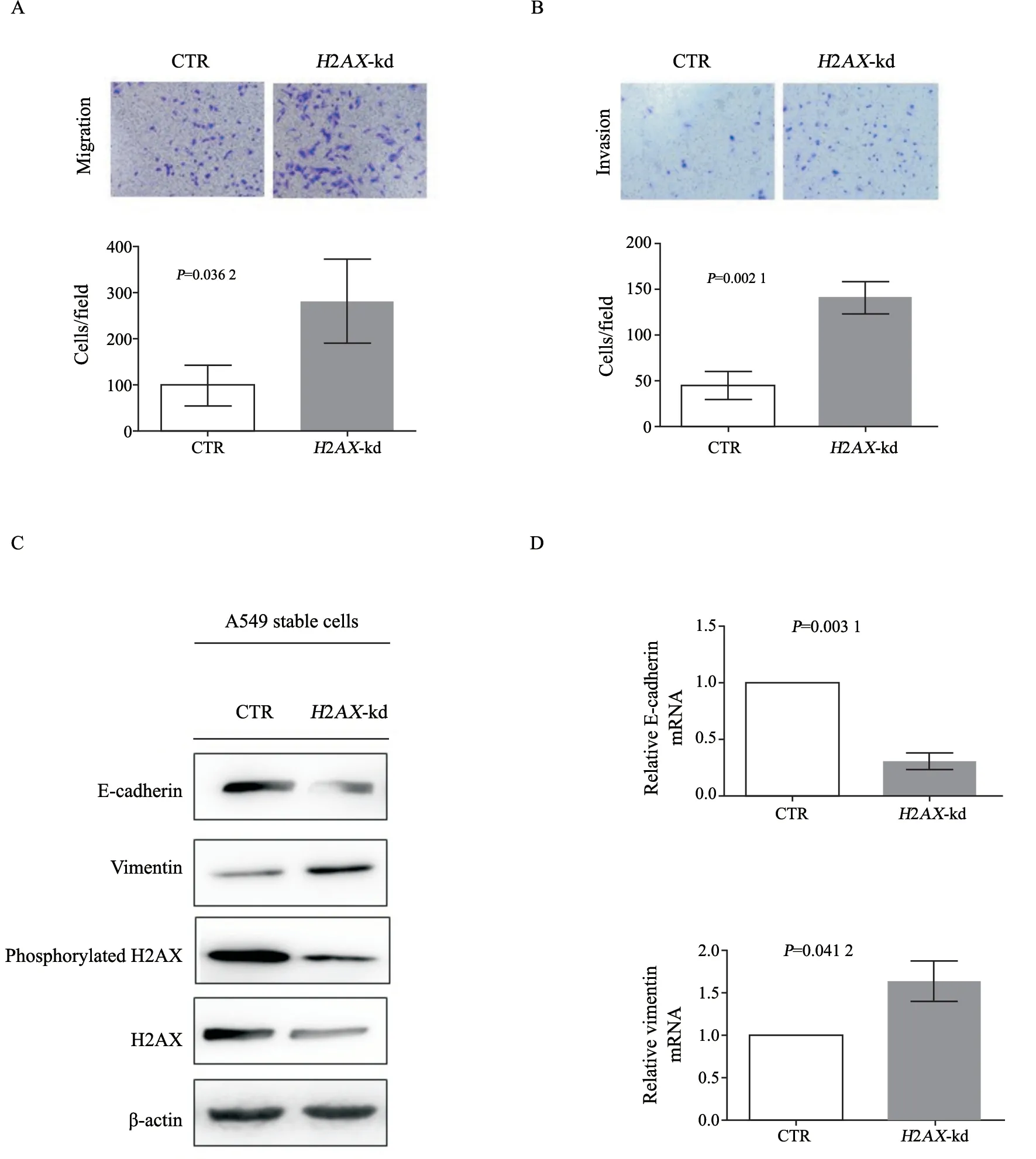
图1 H2AX基因沉默刺激肺癌A549细胞发生EMTFig.1 Knockdown of H2AX stimulated EMT of lung cancer A549 cells
2.2 H2AX Ser139磷酸化参与肺癌A549细胞EMT
目前普遍认为H2AX功能与其磷酸化作用相关[16]。H2AX Ser139磷酸化位点磷酸化修饰后,能够通过表观遗传调控下游基因表 达[17]。为了阐明H2AX抑制EMT是否与其磷酸化有关,我们把本实验室前期制备的肺癌A549稳定株细胞包括vector(转染空质粒)、H2AXwt和H2AX-139m(抑制H2AX磷酸化作用)进行EMT诱导,然后检测过表达H2AX和H2AX-139m基因对肺癌细胞EMT的影响。结果说明,过表达H2AX可上调E-cadherin,下调vimentin蛋白和mRNA水平(图2);H2AX磷酸化位点(Ser139)突变后则相反,下调E-cadherin,上调vimentin蛋白和mRNA水平(图2)。RTFQ-PCR结果与Western blot结果一致。Transwell分析结果也说明过表达H2AX可抑制肺癌细胞的迁移和侵袭能力,H2AX-139m增强A549细胞的迁移和侵袭能力(图3)。以上结果提示肺癌细胞EMT与H2AX磷酸化相关,H2AX磷酸化抑制肺癌细胞EMT。
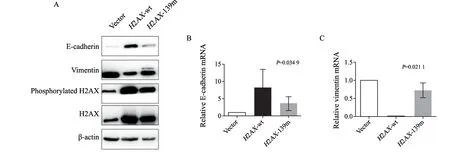
图2 H2AX Ser139磷酸化作用调控E-cadherin和vimentin表达Fig.2 Phosphorylation of H2AX ser139 regulated the expressions of E-cadherin and vimentin
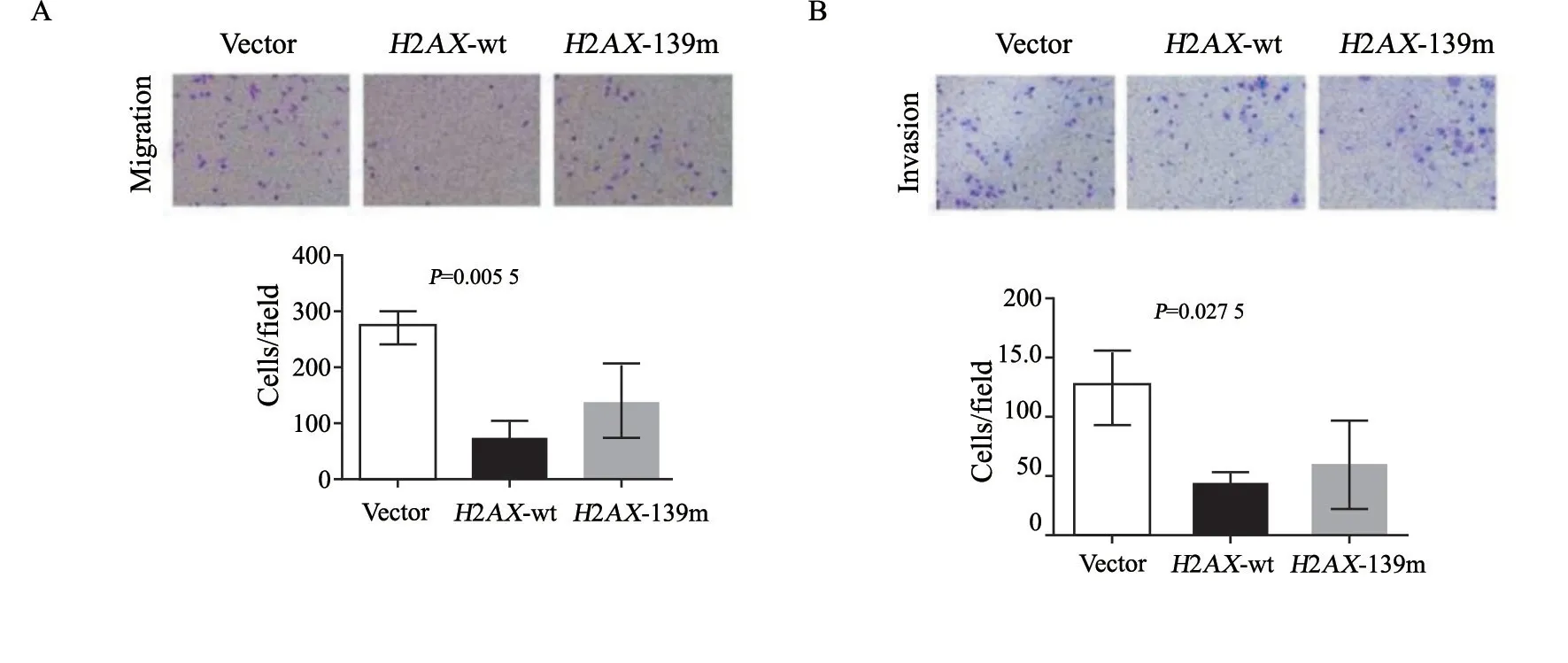
图3 H2AX磷酸化抑制肺癌A549细胞的迁移和侵袭能力Fig.3 Phosphorylated H2AX inhibited the migration and invasion of lung cancer A549 cells
2.3 H2AX磷酸化下调VEGFR-2进而抑制肺癌A549细胞EMT
根据报道,VEGFR-2参与肿瘤EMT的诱发作用[18],目前临床上肿瘤转移治疗主要针对VEGFR-2通路。另外,我们曾经报道组蛋白H2AX表观遗传调控凋亡基因表达[12]。为探讨H2AX是否在转录水平调控VEGFR-2表达,我们把肺癌A549基因沉默稳定株细胞包括H2AXkd和CTR,以及肺癌A549过表达稳定株细胞包括vector、H2AX-wt和H2AX-139m进行EMT诱导,然后提取各种细胞总蛋白执行Western blot检测。结果说明,肺癌A549细胞H2AX表达抑制后刺激VEGFR-2表达;过表达H2AX阻碍VEGFR-2表达,当抑制H2AX磷酸化后则部分消除H2AX对VEGFR-2表达的抑制作用(图4A、B)。检测H2AX基因敲除细胞(H2AX-/-)时获得同样的结果,H2AX基因敲除消除H2AX磷酸化后刺激VEGFR-2表达(图4C)。以上结果 证明H2AX磷酸化下调VEGFR-2。Transwell分析结果说明,肺癌A549细胞转染VEGFR-2 siRNA后则抑制其迁移(图5A)和侵袭能力(图5B),提示VEGFR-2通路确实参与EMT。
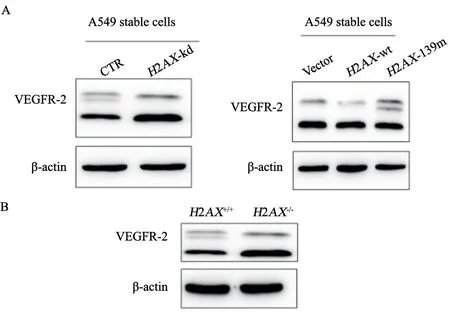
图4 H2AX磷酸化抑制VEGFR-2表达Fig.4 The expression of VEGFR-2 was inhibited by phosphorylated H2AX
我们对VEGFR-2 siRNA的转染效应进行了检测,VEGFR-2 siRNA强烈抑制VEGFR-2的表达(图5C)。以上结果表明H2AX磷酸化下调VEGFR-2进而抑制肺癌A549细胞EMT。
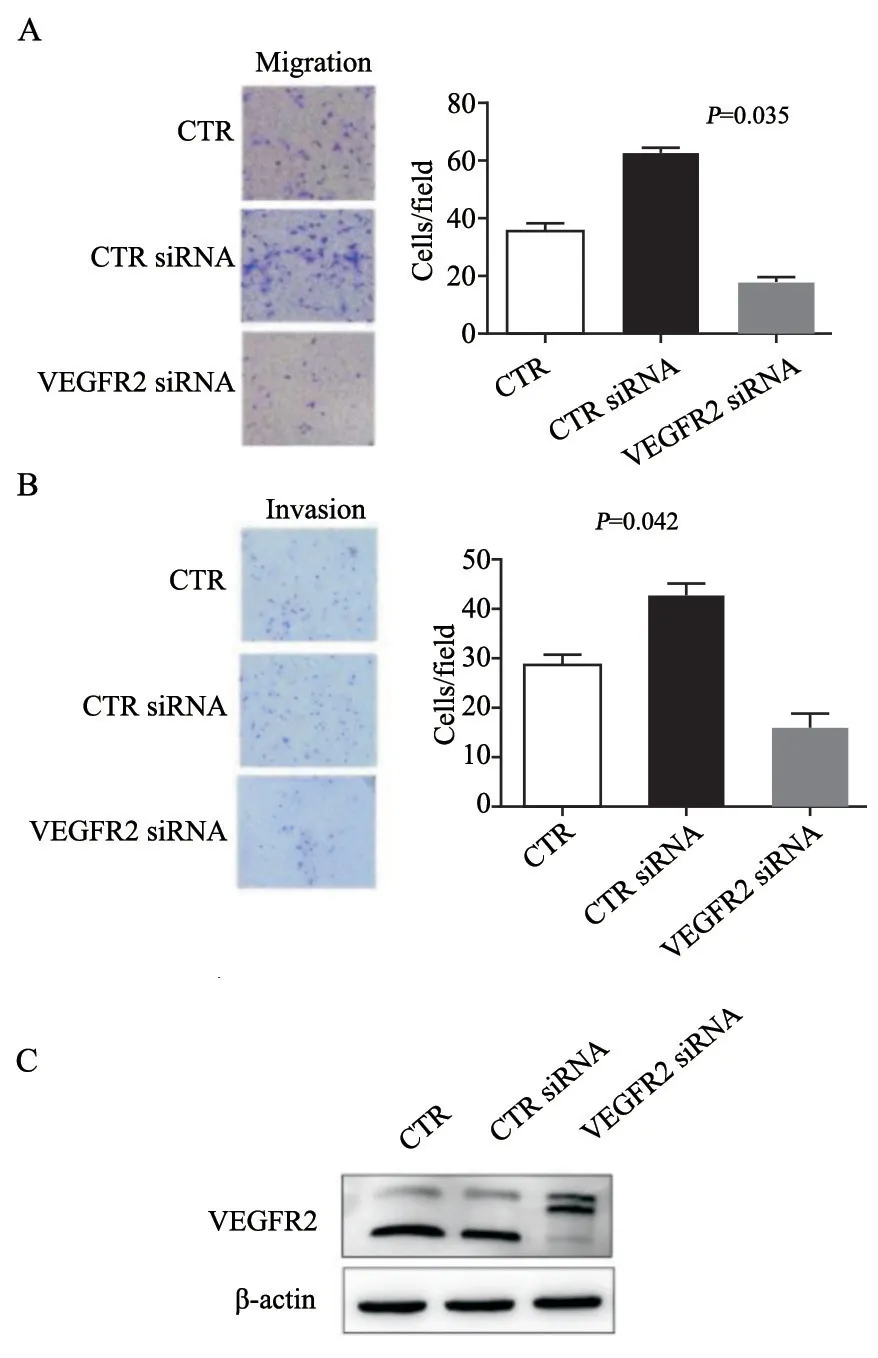
图5 VEGFR-2基因沉默抑制肺癌A549细胞的EMTFig.5 VEGFR-2 knockout inhibited EMT of lung cancer A549 cells
2.4 H2AX磷酸化调控肺癌A549细胞Slug和β-catenin表达
Slug是EMT相关的重要转录因子[19],参与肿瘤细胞的EMT调控。另一种参与肿瘤转移的蛋白β-catenin,主要介导细胞间黏附并参与EMT相关基因表达。我们利用RTFQ-PCR方法验证H2AX磷酸化在转录水平对Slug和β-catenin基因转录作用的影响。结果显示,肺癌A549细胞H2AX表达抑制后(H2AX-kd)上调肿瘤细胞Slug和β-cateninmRNA水平(图6A);过表达H2AX抑制Slug和β-cateninmRNA水平,当阻断H2AX磷酸化后(磷酸化位点突变)则部分消除H2AX对Slug和β-cateninmRNA水平的抑制作用(图6B)。因此,H2AX磷酸化除了调控VEGFR-2表达从而影响肿瘤细胞EMT之外,也参与调控EMT其他相关因子Slug和β-catenin的作用。
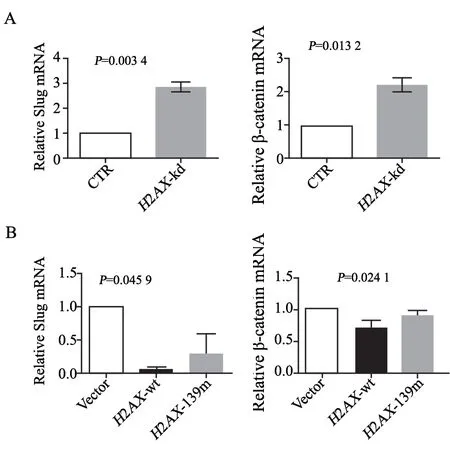
图6 H2AX磷酸化调控EMT相关转录因子Slug和β-catenin的表达Fig.6 Phosphorylated H2AX regulated the expressions of EMT-related transcription factors Slug and β-catenin
3 讨 论
本课题组曾经报道H2AX磷酸化在不同肿瘤细胞中通过不同机制表观遗传调控凋亡基因转录,进而增强肿瘤细胞凋亡敏感性。在白血病细胞中p38MAPK/H2AX磷酸化通路调控凋亡蛋白Bim的表达[14]。另外,我们在肺癌细胞中还发现H2AX磷酸化结合miR-3196启动子并抑制miR-3196表达,进而上调凋亡蛋白PUMA[15]。鉴于H2AX对于肿瘤发生、发展和凋亡至关重要,因此被认为是一种新的抑癌蛋白[20-21]。甚而有人建议,把肿瘤细胞内H2AX水平的变化,作为评估肿瘤患者对化疗药物的敏感性或放疗的耐受性,指导临床治疗。本研究结果首次揭示表观遗传因子H2AX抑制肿瘤新的作用机制,即H2AX磷酸化通过抑制EMT进而抑制肺癌细胞转移。敲低肺癌A549细胞内H2AX表达,则刺激肺癌细胞发生EMT现象包括vimentin上调和E-cadherin下调以及肺癌细胞迁移和侵袭能力的增加;过表达H2AX则抑制EMT;突变H2AX磷酸化位点抑制H2AX磷酸化则部分消除H2AX对EMT的抑制作用。因此,以上结果充分说明H2AX磷酸化抑制肺癌细胞的EMT,为抗肿瘤转移药物研发提供新的靶点分子。至于H2AX磷酸化是否通过诱导肺癌细胞凋亡进而对肺癌EMT的抑制作用产生贡献值得进一步研究。
目前临床上治疗肿瘤转移的主要靶点为VEGF/VEGFR信号转导通路,如VEGF/VEGFR单抗(bevacizumab等)及VEGFR小分子抑制剂(sunitinib等)[18,22],但是VEGFR-2基因表达调控机制不清楚。本实验首次揭示了表观遗传调控蛋白H2AX与VEGFR-2信号转导通路的关系。利用siRNA技术把肺癌A549细胞中的H2AX敲低后,刺激VEGFR-2基因表达;肺癌A549细胞中过表达H2AX则抑制VEGFR-2表达,突变H2AX Ser139磷酸化位点则解除H2AX对VEGFR-2表达的抑制作用。以上结果首次揭示,H2AX磷酸化确实抑制肺癌A549细胞VEGFR-2水平。本研究采用siRNA技术验证肺癌A549细胞内VEGFR-2对EMT的调控作用。结果发现,敲低VEGFR-2则显著抑制肺癌A549细胞的EMT,与报道一致。本实验结果阐明H2AX磷酸化通过下调VEGFR-2进而抑制肺癌A549细胞EMT的分子机制。此外,我们还发现H2AX磷酸化调节肺癌细胞中与EMT有关的转录因子Slug和β-catenin蛋白。由此看来,H2AX调控肺癌细胞EMT的作用机制极其复杂,除了调控VEGFR-2通路外,也参与其他因子的表达调控。
