钴(II)基分子配合物用于光催化二氧化碳还原
2021-06-02张继宏钟地长鲁统部
张继宏,钟地长,鲁统部
天津理工大学新能源材料与低碳技术研究院,材料科学与工程学院,天津 300384
1 引言
随着全球经济发展和能源需求提升,寻找经济和可再生能源是人类21世纪面临的主要挑战之一1,2。目前的能源结构中,85%能源由化石燃料提供。由于化石燃料的不可再生性,将使得其变得更加昂贵且难以获取;另外,化石燃料的使用,排放了大量的二氧化碳(CO2)等温室气体,对全球气候产生严重影响,威胁着人类社会的可持续发展。因此,寻找可替代化石燃料的能源载体非常重要3,4。
受自然界绿色植物光合作用的启发,研究者考虑利用太阳光将CO2转化为燃料或高附加值化学品,这不仅可以降低大气中CO2浓度,缓解环境问题,还可以将太阳能转化成化学能,实现太阳能的存储,解决能源危机5-11。然而,由于CO2的惰性,直接用一个电子将CO2转化为CO2·-非常困难,需要非常负的氧化还原电势(见式(1))12-14;另一种相对有利的途径是通过质子辅助的多电子转移来还原CO2,使CO2可以在相对较正的氧化还原电势下还原为一氧化碳(CO),甲酸(HCOOH),甲醇(CH3OH),甲醛(HCHO),甲烷(CH4)或乙酸(H2C2O4)等(见式(2)-(7))15-18。从热力学上,多质子多电子转移反应有利于CO2还原形成HCHO、CH3OH、CH4等还原产物,但随着质子和电子的增加,动力学势垒通常也将增加,所以将CO2还原成HCHO/CH3OH/CH4等比CO/HCOOH更难19。同时,热力学有利的质子还原反应(-0.41 V vs NHE)还将与CO2还原竞争20,21。因此,开发高效、高选择性的廉价催化剂是实现CO2还原的关键。
光催化CO2还原体系主要分为均相和非均相光催化体系。非均相体系主要是利用具有纳米结构的半导体材料或者金属/半导体复合材料等为催化剂,通过光照,利用价带跃迁到导带的电子直接实现CO2还原。非均相催化剂一般具有稳定的催化性能,也能较好循环利用,但其复杂结构导致催化机理难以揭示。均相光催化CO2还原体系是通过吸光基团吸光和一系列质子和电子转移来实现CO2还原22-27。体系中一般包含分子催化剂(CAT),电子牺牲剂(SD)和光敏剂(PS)。催化反应的第一步是在光照下将PS激发到激发态PS (PS*),被激发后的PS*可以通过两种方式发生猝灭:氧化猝灭和还原猝灭(图1)。在氧化猝灭中,电子从PS*转移到CAT,生成PS+和CAT·-,随后PS+与SD反应,而CAT·-进一步向结合的CO2提供电子实现CO2的还原;在还原猝灭中,激发态的PS*获得SD中的电子,生成强还原剂PS·-和SD+,PS·-将电子转移到CAT形成CAT·-,产生的CAT·-进一步还原结合的CO2分子。虽然反应路径不同,但在这两种途径中都必须要形成CAT·-,因此CAT·-对CO2结合和转化是非常重要的活性中间体。相对于非均相催化剂而言,均相催化剂具有确定的空间结构,有利于结构设计和优化,也有利于研究催化机理和建立构效关系,因而在催化CO2还原方面引起了研究者的广泛关注28。

图1 分子催化剂在光催化CO2还原过程中的两种途径Fig. 1 Two reaction paths of photochemical CO2 reduction in homogeneous catalytic systems.
在均相催化剂中,贵金属Re、Ru、Ir等的配合物均具有较为优异的光催化CO2还原活性,但由于其昂贵的成本将限制其大规模应用,非贵金属Fe、Co、Ni、Mn、Cu等的配合物因而引起了研究者的广泛关注29。Co作为VIII族非贵金属,由于其廉价、储量丰富和多样的氧化态,在非贵金属催化剂用于光催化CO2还原的研究中尤其受到关注30,31。1984年,荷兰乌得勒支大学Tinnemans等人以Co(II)大环配合物作为催化剂32,构建了第一个基于非贵金属催化剂的均相光催化CO2还原体系。随后研究者开展了大量基于不同配体的Co(II)配合物用于光催化CO2还原方面的研究。
本论文主要从配体设计角度出发,介绍Co(II)配合物分子催化剂在光催化CO2还原方面的最新研究进展,重点讨论配体设计和修饰对催化剂效率,选择性和稳定性的影响,总结构效关系。为了更好地分析配体结构对催化效果的影响,我们根据配体类型将钴分子催化剂分为:(1) 基于大环配体钴分子催化剂;(2) 基于吡啶类配体钴分子催化剂;(3) 基于卟啉或卟啉衍生物配体钴分子催化剂;(4) 基于非平面N4配体钴分子催化剂。
2 具有光催化CO2还原活性的钴分子催化剂
2.1 基于大环配体的钴分子催化剂
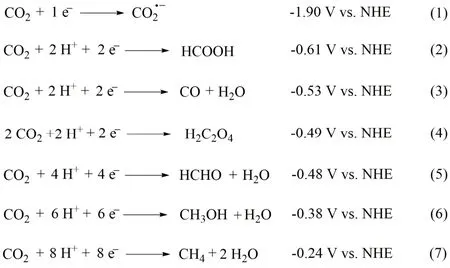
许多配体被用来构建金属配合物并用于催化CO2还原,其中大环配体能够稳定催化循环中形成的中间体,从而提高催化活性。1984年,荷兰乌得勒支大学Tinnemans等人使用三种四氮杂大环配体合成系列Co(II)配合物(C1-C3;图2),它们可作为分子催化剂在水溶液(pH 4.0)中实现光催化CO2还原。以[Ru(bpy)3]2+作为光敏剂和抗坏血酸盐(HA-)作为电子牺牲剂,C1和C2光催化CO2还原成CO的选择性分别为22%和32%,C3对CO选择性低于1% (表1,Entries 1-3)32。在光照18 h后,C1和C2的TONCO分别为22和9.6。作者提出的催化机理是:抗坏血酸盐使激发态的*[Ru(bpy)3]2+发生还原猝灭生成还原性更强的[Ru(bpy)3]+(图3,步骤1和2),然后[Ru(bpy)3]+将[CoIIL] (L代表C1-C3的配体)还原成[CoIL] (图3,步骤3)。在质子溶剂中,[CoIL]与质子结合生成[CoIIILH]-(图3,步骤4),接下来可能经历两种不同的途径:一种是[CoIIILH]-与另一质子反应生成H2(途径a),另一种是CO2插入Co―H键形成[CoIIILCOOH],然后结合一个质子释放出CO和H2O(途径b)。最后[CoIIIL]与抗坏血酸盐反应,被还原成[CoIIL] (图3,步骤5),完成一个催化循环。美国Fujita教授等人随后用瞬态光谱证明了光催化反应中中间体[CoIL]和相应的[CoL(CO2)S] (S = 溶剂)的形成33-36,说明在CO2存在的情况下,途径b是形成CO的有效路径。
图2 基于大环配体的钴分子催化剂Fig. 2 Chemical structures of Co molecular complexes based on macrocyclic ligands.
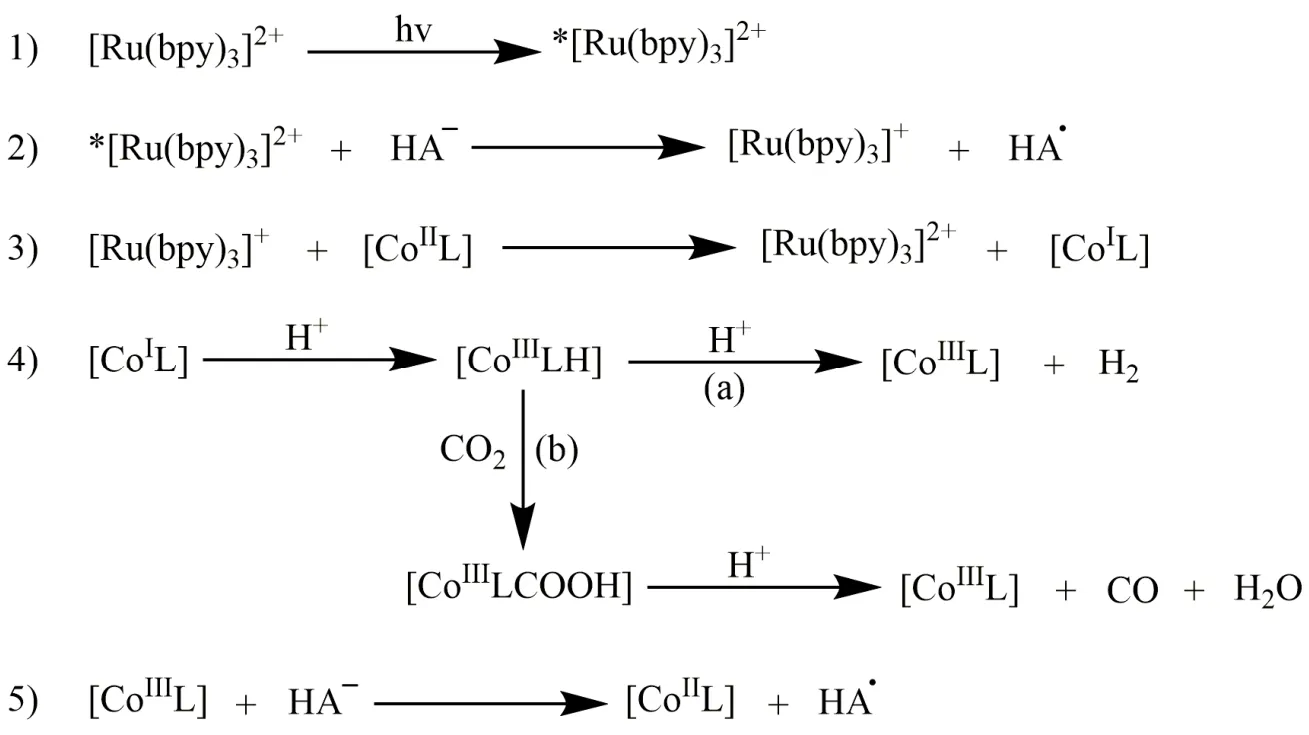
图3 Tinnemans等人提出的CoIIL配合物光催化还原CO2为CO机理Fig. 3 Proposed mechanism of photochemical reduction of CO2 to CO by Tinnemans et al.
1995年,美国Yanagida和Fujita等报道了另一Co(II)大环催化剂(C4;图2)34,以三联苯(pterphenyl)作为光敏剂,三乙醇胺(TEOA)作为电子牺牲剂,C4在CH3CN/MeOH混合溶剂体系中可以光催化还原CO2为CO和甲酸,C4表现出良好的选择性和高量子产率(表1,Entry 4)。当用吩嗪(phenazine)作为光敏剂和三乙胺(TEA)作为电子牺牲剂时,CO2还原成甲酸的选择性明显提高(>90%),量子效率为0.07 (表1,Entry 5)35。此外,他们进一步探究了C4催化剂中大环配体的结构对催化活性的影响(表1,Entries 6-10)36,37。他们发现:在相同条件下,带有更多甲基取代基的C6光催化CO2还原的活性明显比C5的低(表1,Entries 6和7);将C4中四氮杂大环配体中N-甲基化后形成的C7不能实现对CO2的还原(表1,Entries 8和9)。这些实验结果表明,太多甲基取代基可能会导致位阻太大,阻碍催化中心与CO2结合,从而影响CO2还原催化活性。他们还发现没有N―H键的四氮杂大环Co(II)配合物(C9;图2)光催化CO2还原活性比C8更低(表1,Entry 10)。除N4大环外,研究者还设计合成了基于N5大环的钴分子配合物(C10;图2),发现以fac-Ir(ppy)3为光敏剂和TEA为电子牺牲剂时,它能光催化还原CO2为CO,TON达到270,对CO的选择性达到97% (表1,Entry 11)38。有意思的是,当催化中心换成Fe后,在相同条件下,CO2的还原产物为甲酸盐。
2.2 基于吡啶基配体的钴分子催化剂
吡啶类化合物是合成钴分子催化剂的常用配体,将Co(II)与联吡啶(bpy)配位形成的[Co(bpy)3]2+(C11;图4)和双金属配合物C12和C13 (图4),在光照条件下能将CO2还原为CO,但它们的TON和选择性都较低(表1,Entries 12-14)39,40。2016年香港城市大学Lau和法国Robert等人报道了钴与四联吡啶形成的催化剂C14 (图4),在[Ru(bpy)3]2+和1,3-二甲基-2-苯基-2,3-二氢-1H-苯并[d]咪唑(BIH)的CH3CN/TEOA体系中,光催化CO2还原成CO的TON高达2660,选择性为98%,量子效率(ΦCO)达6.74% (表1,Entry 15)41。用吡啉(purpurin)替换[Ru(bpy)3]2+作为光敏剂,该催化剂仍表现出较高的活性,TON达790 (表1,Entry 16)。随后Chen, Lau和Robert等人进一步合成了一种基于四联吡啶双核钴催化剂(C15;图4)42,他们发现C15在不同pH值溶液中能将CO2选择性的还原成HCOO-或CO。在弱碱性乙腈溶液中,CO2的还原产物为甲酸,TON达到821,选择性为97% (表1,Entry 17);在弱酸条件下,CO2的还原产物为CO,TON达到829,选择性达99% (表1,Entry 18)。机理研究表明催化过程由两个Co(II)协同作用完成。2018年,Fujita课题组合成了两个具有五齿聚吡啶配体的Co(II)配合物(C16和C17;图4)43。当用C16作为催化剂,[Ru(bpy)3]2+作为光敏剂,TEA作为电子牺牲剂(表1,Entry 19),在可见光照射下,CO2可被还原成CO和HCOOH。有趣的是,在配体的末端吡啶上接上甲氧基后(C17;图4),CO2还原产物只有CO (表1,Entry 20)。
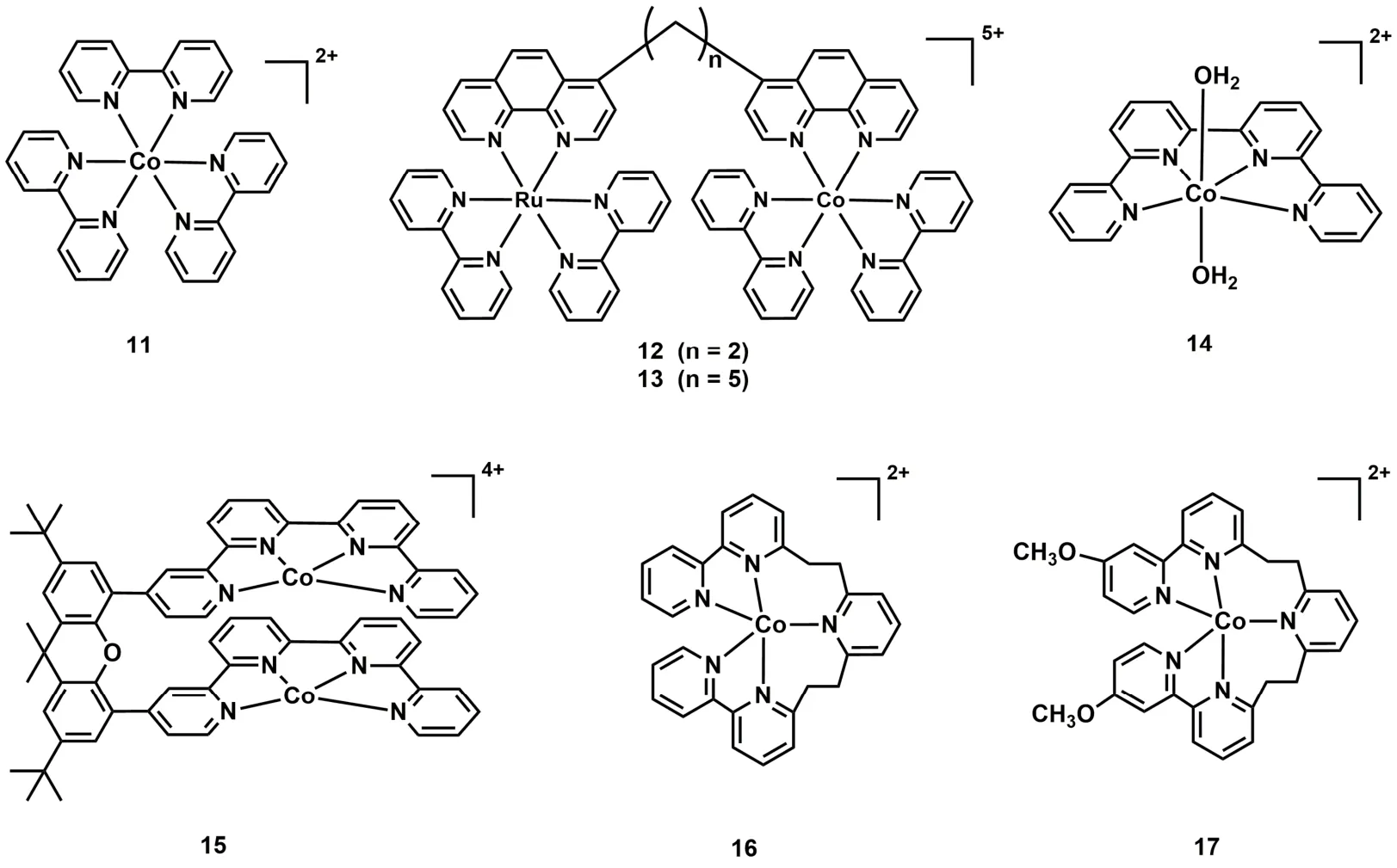
图4 基于吡啶基配体的钴分子配合物Fig. 4 Chemical structures of Co molecular complexes based on polypyridyl ligands.
2.3 基于卟啉或卟啉衍生物配体的钴分子催化剂
卟啉及其卟啉衍生物具有良好的吸光性能,基于卟啉及其卟啉衍生物配体的金属配合物被广泛用于光催化反应。1998年,美国国家标准技术研究院Neta等人发现用三乙胺作为电子牺牲剂,在紫外可见光(λ > 320 nm)照射下,钴卟啉(C18;图5)在乙腈溶剂中能将CO2还原成CO和甲酸44。光照200 h后,产生CO和甲酸的总TON超过300 (表1,Entry 21)。作者通过电化学和光谱分析捕捉到了反应的活性中间体[Co0P] (P = 卟啉)。此外,Neta等人进一步研究发现将三联苯作为光敏剂和三乙胺作为电子牺牲剂时,在紫外可见光的照射下,卟啉钴衍生物(C19和C21;图5)和酞菁钴(C20;图5)也能将CO2还原成CO或者甲酸(表1,Entries 22-24)40,45,46。
2019年,日本九州大学Sakai课题组合成了两种水溶性的钴卟啉(C22和C23;图5)。在可见光下,它们能够高效还原CO2为CO,当以[Ru(bpy)3]2+为光敏剂和抗坏血酸盐为电子牺牲剂时,C22能在水溶液中将CO2还原成CO,TON高达926,选择性大于82% (表1,Entry 25)47。当作者用廉价的Cu(I)配合物(CuPS)48替代[Ru(bpy)3]2+作为光敏剂时,在纯水体系中,C22也具有优异的光催化还原CO2为CO性能,TONCO达到1085,选择性达到90% (表1,Entry 26)49。在同样的条件下,C23表现出更为优异的催化性能,将CO2还原成CO的TON高达2680,TOF达到2600 h-1(表1,Entry 27)49。
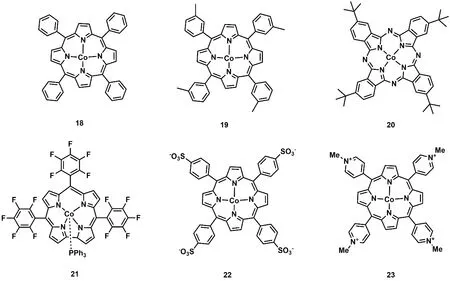
图5 基于卟啉或卟啉衍生物配体的钴分子配合物Fig. 5 Chemical structures of Co molecular complexes based on porphyrin and porphyrin-like ligands.
2.4 基于非平面N4配体的钴分子催化剂
除平面结构的Co(II)配合物外,研究者还探究了基于非平面N4配体Co(II)配合物光催化CO2还原性能。2015年,香港理工大学Chan等人报道了一种含有四齿三脚架配体的Co(II)配合物[Co(TPA)Cl] (TPA = 三(2-吡啶基甲基)胺) (C24;图6)50-52。使用Ir(ppy)3作为光敏剂和TEA作为电子牺牲剂,C24在乙腈中能够催化CO2转化为CO,选择性达到85%,TONCO达953 (表1,Entry 28)。随后,华中科技大学王峰等人将TPA配体进行修饰,合成了一种顺式双核Co(II)配合物(C25;图6)53。光催化CO2还原结果显示C25能将CO2还原成CO和HCOOH (表1,Entry 29)。DFT计算结果表明:活性中心配位点上的氧原子在CO2转化为CO和HCOOH都起着重要作用。
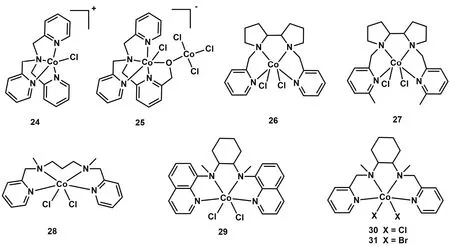
图6 基于非平面N4配体的钴分子配合物Fig. 6 Chemical structures of Co molecular complexes based on nonplanar N4 ligands.
2016年,香港大学支志明课题组合成了一系列新型的顺式[Co(N4)Cl2]配合物(C26-C29;图6),用于光催化CO2还原54。在这些配合物中,活性中心Co(II)与配体上四个氮原子和两个氯离子配位,形成六配位八面体构型。光催化实验结果表明:配合物中N4配体的不同结构极大地影响了它们的光催化CO2还原活性(表1,Entries 30-33)。其中,以Ir(ppy)3作为光敏剂和TEA作为电子牺牲剂,C26表现出最高的光催化CO2还原活性和选择性,可见光照射60 h,TONCO达到368,选择性为95% (表1,Entry 30)54,但在C26的配体吡啶环上修饰上甲基,形成的配合物C27却不能实现对CO2的光还原(表1,Entry 31)。作者还合成了两个类似配体的Co(II)配合物C28和C29,实验结果表明:C28能将CO2还原成CO,但TONCO只有13,而C29对CO2还原却没有活性(表1,Entries 32-33)。由此可见配体中部分基团的空间位阻效应对催化活性具有重要影响。电化学和光谱研究表明,激发态Ir(ppy)3的电子转移到C26所产生的CoI是CO2结合的活性物种。DFT计算表明,配位的Cl-对催化活性也有显著影响,与不存在Cl-的相比,[Co(CO2)LCl]0质子化得到[Co(COOH)LCl]+是放热的过程,有助于CO2催化还原过程的进行。此外,华中科技大学王峰等人系统研究了不同卤素离子对催化活性的影响,他们合成了两个结构相似的Co(II)配合物(C30和C31)55,56。如图6所示,这两个配合物具有相同的N4配体和金属中心,但配位的卤素离子不同,与C30催化中心配位的是Cl-,而C31的是Br-,在相同的条件下,它们表现出不同的催化CO2还原活性(表1,Entries 34-37)。在光照12 h内,C31(TONCO= 102)的CO生成速率比C30 (TONCO= 56)更快,说明卤素对CO的形成有影响。但是光照48 h之后,由于C30稳定性更高,C30的TON(TONCO= 470)高于C31 (TONCO= 403)55。电化学和光谱研究表明,C31光生电子转移(PET)过程的速率常数和自由能变化比C30更大,与催化实验结果一致。
四齿三脚架配体通常与金属结合形成五配位的单核金属配合物57-59。在催化过程中,轴向位置的溶剂分子可以解离,暴露出空位点键合底物分子。因此,基于四齿三脚架配体的金属配合物也可能具有光催化CO2还原的能力。最近我们组通过配体修饰合成了一系列基于四齿三脚架配体的Co(II)配合物分子催化剂,并构建了稳定高效的光催化CO2还原体系。最初,我们采用具有富电子的苯并咪唑基团的四齿三脚架配体NTB (NTB = 三-(苯并咪唑基-2-甲基)胺)与Co配位形成单核Co(II)配合物(C32;图7)58,使用[Ru(phen)3](PF6)2作为光敏剂和TEOA为电子牺牲剂,在含水溶剂(CH3CN/H2O,v/v = 4 : 1)中,C32光催化CO2还原成CO的TON达到1179,选择性为97% (表1,Entry 38)58。随后,我们研究了空间位阻对光催化CO2还原活性的影响,设计了两个基于三脚架配体结构相似的配合物C33和C3460,如图7所示,C33中催化中心Co(II)周围的是三个异丙基;C34中的是苯甲基,光催化实验结果表明:C33光催化CO2还原的催化效率是C34的7倍(表1,Entries 39和40),这可能是由于C33中异丙基比C34的苯甲基空间位阻更小,有利于CO2与C33中的催化中心CoII结合以及激发态的光敏剂与催化剂C33之间的电子转移33,34,61。我们进一步研究了共轭效应对光催化CO2还原活性的影响,设计合成了三种具有不同共轭基团的三脚架配体及其相应的Co(II)配合物(C35-C37;图7)62,实验结果表明含有蒽甲基的C35和含有萘甲基的C36光催化CO2还原成CO的活性和选择性都比C37高(表1,Entries 41-43)。对照实验和DFT计算表明,C35和C36具有更优异的催化性能是由于配体中的共轭取代基使得催化中心CoII具有更正的还原电势,同时,共轭基团也促进了激发态光敏剂分子上的电子往催化剂分子上转移,从而显著提高了CO2还原成CO的效率。此外,我们还研究了催化中心Co(II)的配位环境对光催化CO2还原活性的影响(C38-C41;图7)63,发现随着喹啉基团数量的逐个增加,配合物的光催化活性逐步提升,最高TON达到10650,选择性也接近100%。C41催化活性略低,可能原因是喹啉基团的增加,导致催化中心周围空间位阻增大,不利于Co(II)与底物CO2作用(表1,Entries 44-47)。
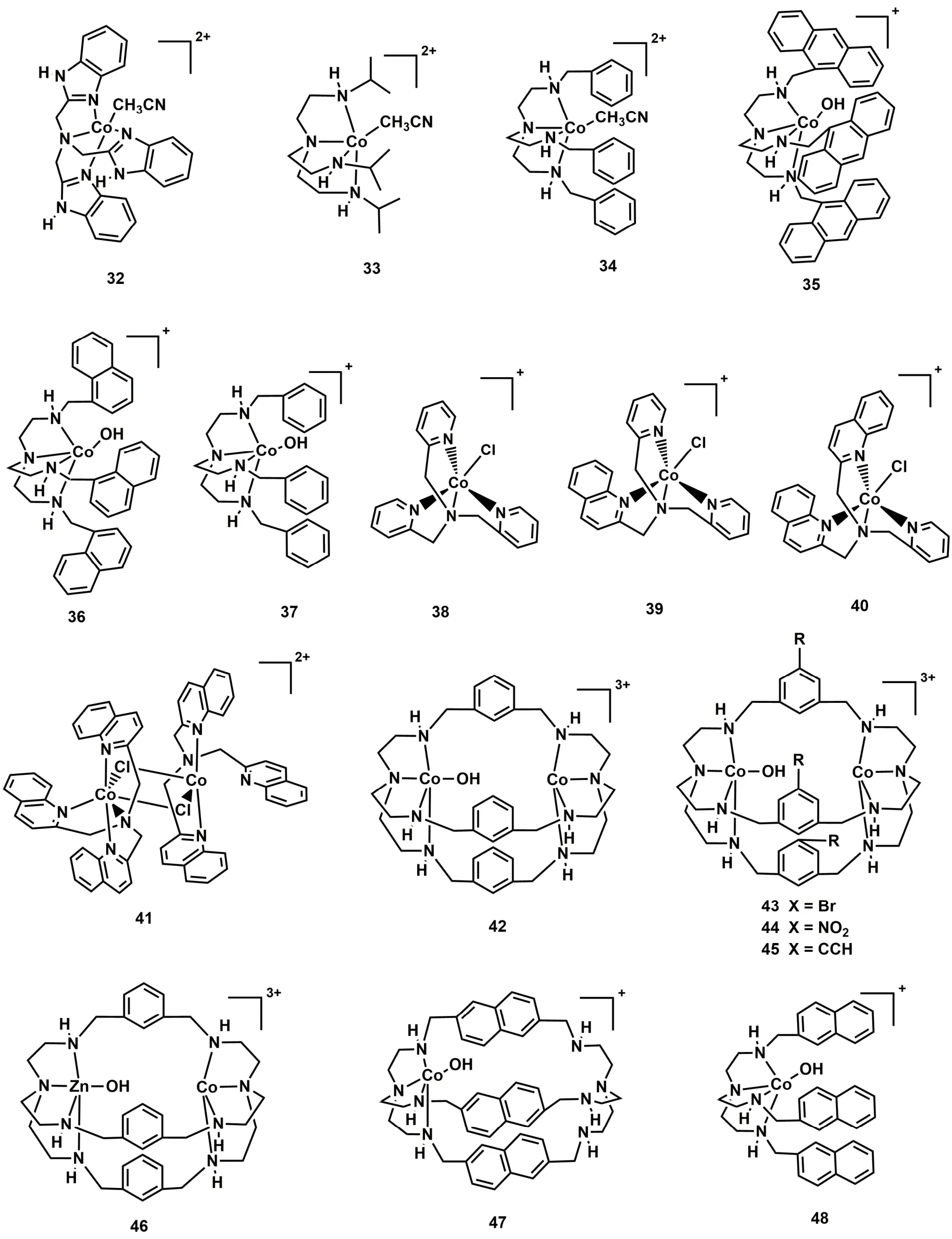
图7 基于非平面N4配体的钴分子配合物Fig. 7 Chemical structures of Co(II) molecular complexes based on nonplanar N4 ligands.
在研究单核Co(II)配合物光催化CO2还原为CO时,我们发现中间体[O=C―OH]中C―O键的断裂是反应的速控步,需要较高的反应能垒,含有双中心的双核金属配合物可能有助于中间体中C―O键的断裂,加快催化反应的进行,提升催化效率。根据这个设想,最近我们合成了一个基于氮杂穴醚大环配体双核Co(II)配合物(C42)64,65。光催化实验结果表明:C42光催化还原CO2为CO的TONCO高达16896,选择性也达到98%(表1,Entry 48),催化活性远远高于相应的单核Co(II)配合物(C34;表1,Entry 40),从实验上证明了两个Co(II)金属中心协同催化作用的存在。接着我们进一步通过理论计算提出了双核金属协同催化机理:一个Co(II)作为催化活性中心结合和还原CO2,另一个Co(II)作为辅助催化位点促使中间体[O=C―OH]中C―O的断裂和―OH的离去,协同促进了CO2向CO迅速转化,并由此提出了双核金属协同催化CO2转化为CO的反应路径(图8)。首先,C42快速结合CO2形成碳酸根桥联的双核Co(II)配合物C42a,然后进行质子耦合电子转移反应生成C42b,C42b结合一个质子并脱去一分子H2O得到C42c。C42c通过过渡态TS1-1转化为C42d,C42d经历第二次质子耦合电子转移反应得到C42e,C42e经过TS2-1形成C42f,在这一步中间体[O=C―OH]中C―O通过两个Co(II)协同作用发生断裂,最后释放出CO完成催化循环66。这是首次利用双核金属协同催化实现光催化CO2还原。我们进一步研究发现,在电催化条件下双核金属分子催化剂也存在协同催化作用,极大地提升了CO2还原活性67。随后葡萄牙里斯本大学Martinho等人对C42进行修饰,合成了三种苯环上具有不同取代基的双核Co(II)配合物C43-C4568,实验结果表明催化剂在低浓度和较短照射时间下,带有给电子基团炔基的配合物C45表现出最高的催化活性,TONCO达到14210 (表1,Entries 49-51)。随着反应时间的延长,生成的CO会进一步反应生成CH4,其中带有―Br取代基的配合物C43产甲烷的活性最高(表1,Entries 52-54),理论计算结果表明C43能改变键能,以及使反应中间产物HCO3-更牢固地与催化剂结合,从而表现出较高的活性。
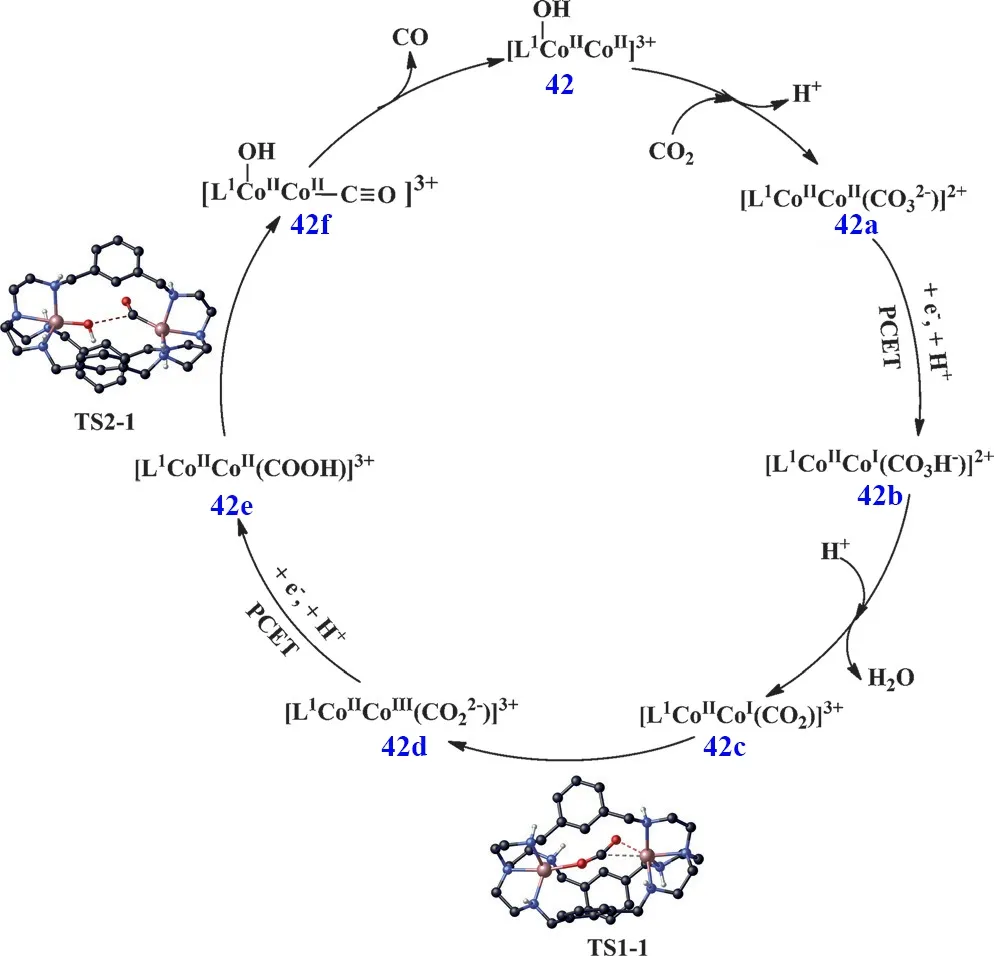
图8 C42可见光驱动CO2还原为CO的催化机理Fig. 8 Catalytic mechanism of C42 for the visible-light driven reduction of CO2 to CO.
为了进一步验证和利用双金属协同催化,我们将C42中的一个CoII换成ZnII合成了一种双核异金属配合物C4669,通过光催化CO2还原实验我们发现,在相同条件下,双核异金属配合物C46的TONCO达到65000 (表1,Entry 55),远远高于C42(表1,Entry 48),是C42的4倍,选择性也达到98%。C46催化活性的增强归因于CoII与ZnII之间增强的协同催化效应,因为Zn(II)对OH-具有更强的结合力,更有助于反应中间体[O=C―OH]的C―O的断裂,极大地促进了CO2向CO的转化。除此之外,我们还设计合成了另一种穴醚配体,希望通过调节两个金属中心之间的距离从而进一步研究双核金属协同催化CO2还原,遗憾的是我们只得到了单核Co(II)配合物C4770。为了研究穴醚配体的限域作用,我们还合成了另一个相应的三脚架配合物C48。实验结果发现,C47比C48具有更高的光催化还原CO2为CO的活性(表1,Entries 56和58)。即使在模拟烟道气氛围中CO2/Ar = 1/9 (v/v),C47光催化CO2还原为CO仍表现出高活性和选择性(表1,Entry 57)。对照实验和DFT计算结果表明C44更高活性归因于穴醚配体的限域作用,这使活性中心CoII拥有更正的还原电位和更低的过渡态能垒。除双核Co(II)配合物,最近日本大阪大学Masaoka等人合成了一个五核Co(II)配合物(C49)71,用Ir(ppy)3作为光敏剂,BIH作为电子牺牲剂,在DMA和TFE混合溶剂中,光催化CO2还原成CO和甲酸的TON为58.4和55.7 (表1,Entry 59)。
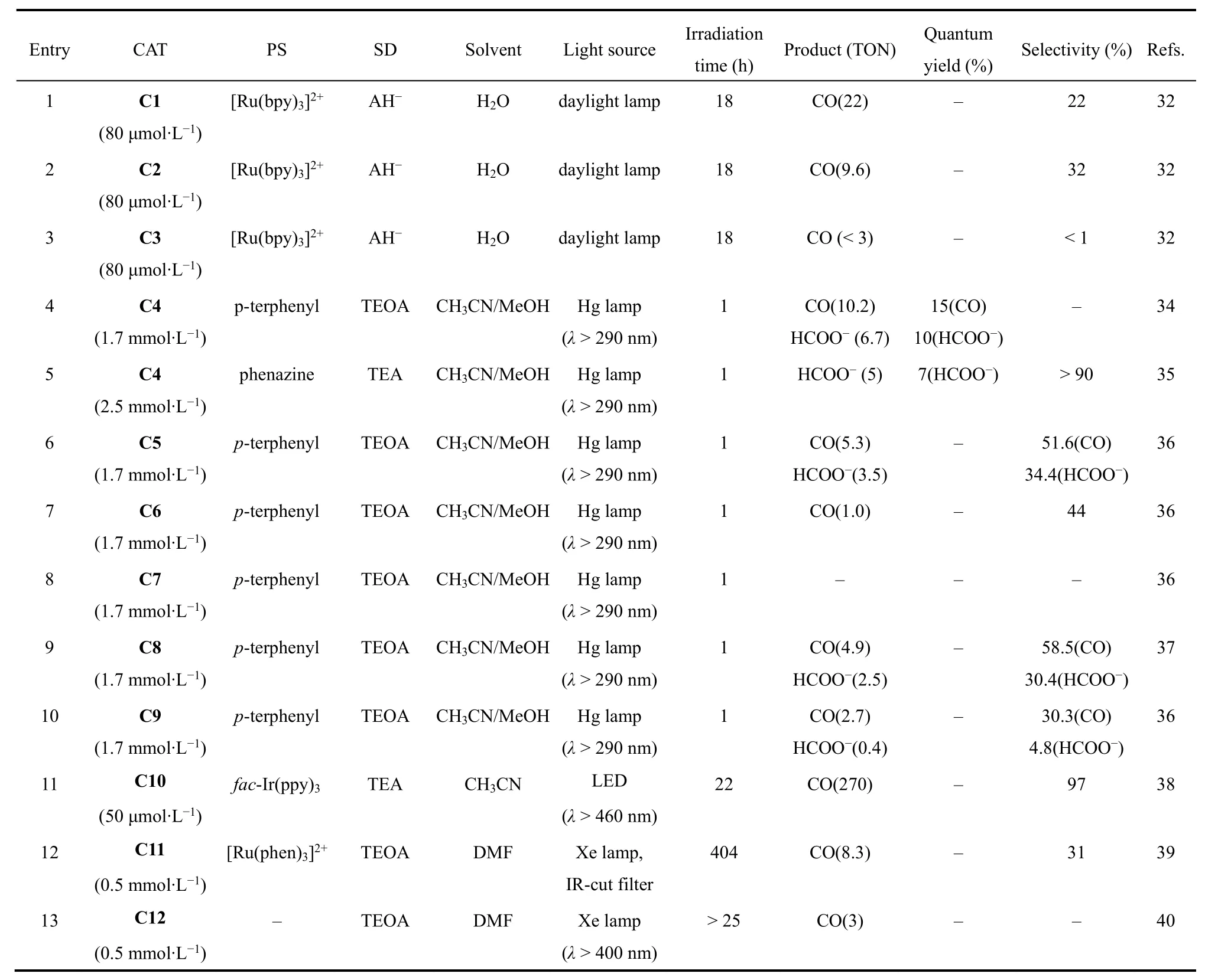
表1 Co(II)配合物分子催化剂光催化CO2还原Table 1 Co(II) complexes as molecular catalysts for photocatalytic CO2 reduction.
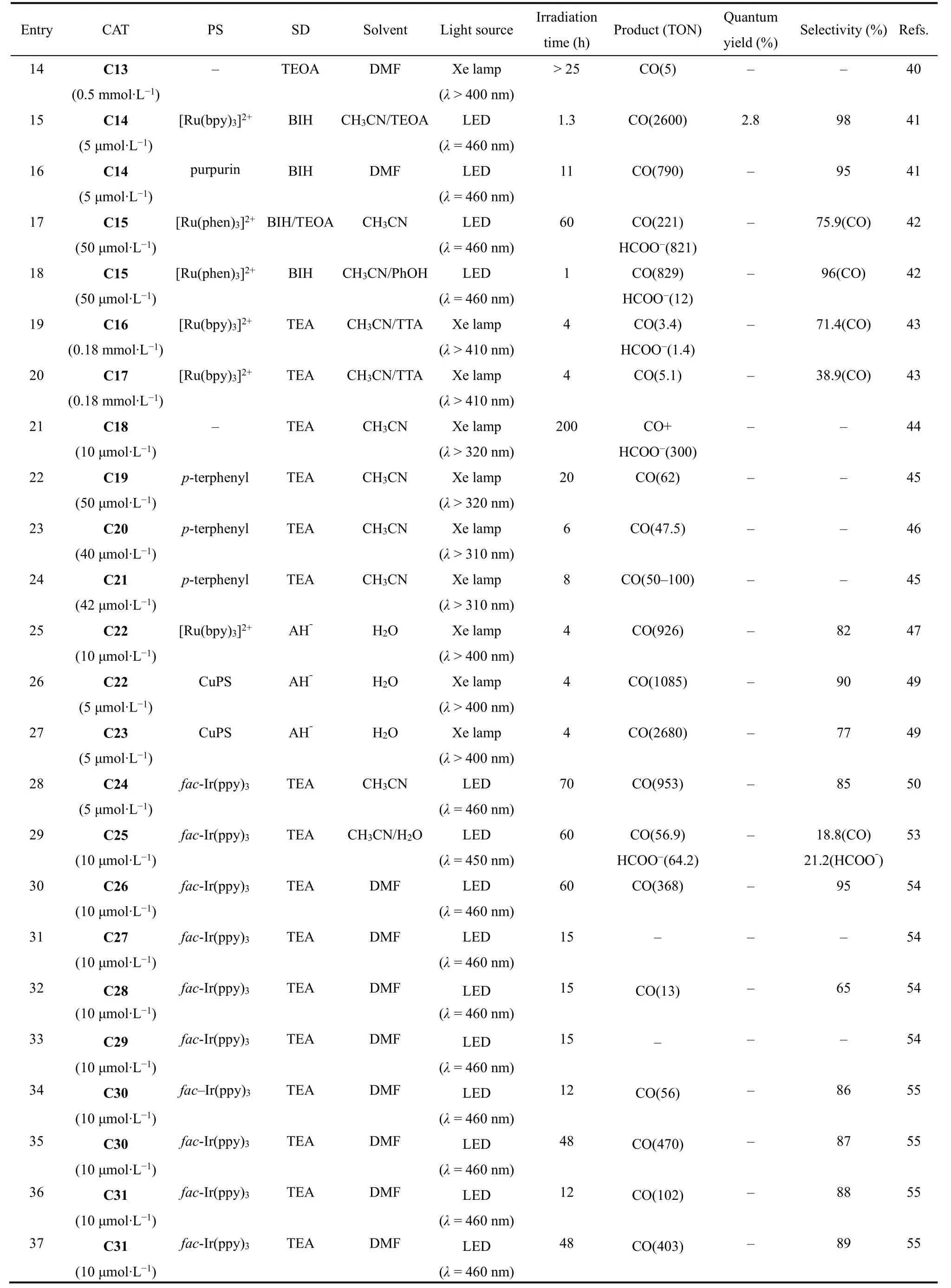
continued Table 1

continued Table 1
3 具有光催化CO2还原活性钴分子催化剂的异相化
为了提高分子催化剂的稳定性,以及减少对贵金属光敏剂的依赖,研究者也开展了许多将分子催化剂与无机半导体材料如氮化碳、石墨烯、二氧化钛和硅等结合起来制备光催化CO2还原异相催化剂的工作。2014年印度石油研究所Jain等人合成一个卟啉钴分子催化剂C50(图9)72,C50上氨基与氧化石墨烯(GO)上的羧基通过席夫碱反应形成C50-GO催化剂,实验结果表明以TEA作为电子牺牲剂,光照48 h,C50-GO能将CO2还原成甲醇,反应速率达到78.8 μmol·g-1·h-1,远远高于单纯的GO或GO与C50物理混合作为催化剂的催化活性。2016年日本大阪大学Fukuzumi等人利用π-π堆积作用将C51 (图9)与碳纳米管(CNTs)复合形成C51-CNTs催化剂。以[Ru(Me2phen)3]2+作为光敏剂,TEA作为电子牺牲剂,在CH3CN和H2O溶液中,C51-CNTs能高效光催化CO2还原成CO,TONCO达到50173。2017年天津大学叶金花教授将带有羧基的卟啉钴(C52;图9)通过共价键连接的形式与g-C3N4形成C52-g-C3N4复合催化剂74,用TEOA作为电子牺牲剂,在乙腈溶液中不添加其他光敏剂的条件下,CO的生成速率达到17 μmol·g-1·h-1。后续研究者报道了一系列钴分子配合物(C53-C56;图9)通过共价键或超分子作用与g-C3N4连接起来,并用于光催化CO2还原的研究工作75-78。其中C53和C54分别与g-C3N4形成的复合催化剂光催化CO2还原成CO的TON分别达到90和128,C55-g-C3N4光催化成CO的生成速率达到8.1 μmol·h-1。而C56-g-C3N4能将光催化CO2还原成甲醇,产率最高可达538.7 μmol·g-1·h-1(以C56的量计算)。最近我们组将带负电的CdS量子点和带正电的双核钴分子配合物(C57;图9),通过静电作用组装成C57-CdS79,用TEOA作为电子牺牲剂,在NaHCO3水溶液中,光催化CO2还原成CO的TON达到1380。这些异相化后的钴基催化剂,均能表现出良好的循环稳定性。
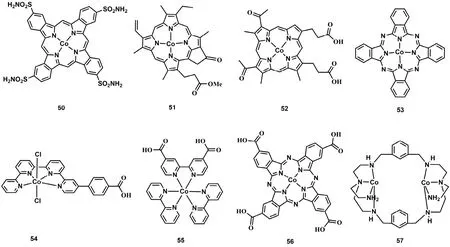
图9 用于异相化的钴分子配合物Fig. 9 Chemical structures of Co(II) molecular complexes used for heterogeneity.
4 总结和展望
在本文中,我们主要从配体角度综述和讨论了非贵金属钴分子催化剂在光催化CO2还原方面的最新研究进展。从总结的研究工作可以发现,配体结构对钴分子催化剂催化CO2还原性能具有重要影响,配体修饰能明显改善催化剂的催化活性。但是,目前均相光催化CO2还原离实际应用还有很长的路要走,还存在一些关键问题没有解决。首先,当前报道的光催化CO2还原分子催化体系,除了我们课题组发现的一个普适性含水催化体系外18,58,80,其它几乎都需要在无水体系中进行。因为在含水体系中,水的质子还原竞争反应将极大降低CO2还原的选择性。因此,要实现真正人工光合作用,首先要解决的问题是开发在含水或全水体系中高效高选择性的CO2还原分子催化剂。其次,最理想的人工光合作用是在太阳光和催化剂作用下,CO2还原反应与水氧化反应相耦合,实现真正的人工光合作用。然而,已报道的分子催化剂在光催化CO2还原时需要加入电子牺牲剂,而大部分电子牺牲剂的成本比CO2的还原产物如CO、CH4、CH3OH等还高,从而无法在实际中得到应用。因此,开发新的分子催化体系,利用水氧化产生的电子还原CO2,实现真正的人工光合作用,是光催化CO2还原分子催化剂的一个极其重要而又富有挑战的发展方向。
尽管目前报道的包括Co(II)配合物在内的分子催化剂,均需要在使用电子牺牲剂的情况下才表现出光催化CO2还原活性,而且大多数催化剂反应活性、产物选择性和稳定性仍然不够理想,但是,这种模拟光合作用半反应的催化体系,对筛选催化剂,分析和总结催化剂的构效关系,以及揭示催化反应机理,仍具有重要的研究意义。只有对催化剂的构效关系和催化机理有深入的认识,才有可能指导设计出更加优异光催化性能的CO2还原分子催化剂。为了获得理想的光催化CO2还原分子催化剂,我们认为在设计催化剂时,以下几个方面值得考虑:(1) 构建双核或多核Co(II)配合物催化剂,利用双核金属间的协同催化作用,提高催化剂对光催化CO2还原的活性和选择性。(2) 配位原子合适的Lewis碱性。理论上,配位原子的弱Lewis碱性会使Co(II)更容易被还原,获得更正的氧化还原电势,但弱Lewis碱性也可能使配体与Co(II)结合不牢固,导致Co(II)基分子催化剂不稳定。(3) 配体上修饰有效的功能基团。这些功能基团最好能一方面富集CO2分子,使Co(II)催化中心周围具有高浓度的CO2,从而加强Co(II)与CO2分子的接触,提高催化效率。另一方面可以与催化中心结合的反应中间体产生超分子作用,对反应中间体有一定的稳定作用,使其沿着预期的反应方向进行。有机功能基团―OH,―NH2等既能作为氢键给体和受体与反应中间体形成氢键作用,又能与CO2分子作用富集CO2,因此,在催化剂设计中,可以考虑在配体的合适位置修饰―OH,―NH2等功能基团,提高光催化CO2还原效率。总之,Co(II)分子配合物在光催化CO2还原方面展现了潜在的应用前景,也取得了较大进展,虽然目前还存在活性、稳定性和量子效率普遍比较低、多碳产物少等一些问题,但通过对催化机理的深入理解和揭示,进一步总结和积累构效关系,可以指导设计出具有优异光催化CO2还原性能的Co(II)基分子催化剂。
