大黄标准汤剂量值传递规律研究
2021-05-27窦志华周荣荣周云中王兆龙倪丽丽
戴 莹,施 凯,窦志华,,周荣荣,周云中,王兆龙,倪丽丽
1.南京中医药大学,江苏 南京 210023
2.南京中医药大学南通中西医结合临床医学院,江苏 南通 226006
3.南通市第三人民医院,江苏 南通 226006
4.精华制药集团股份有限公司,江苏 南通 226005
中药标准汤剂同时是配方颗粒和经典名方制剂的物质基准[1],其核心作用是作为两者工艺研究和质量评价量值传递的桥接。国家药品监督管理局2021年1月发布的《中药配方颗粒质量控制与标准制定技术要求》明确提出:为使中药配方颗粒能够承载中药饮片安全性、有效性,需要以标准汤剂为桥接,该标准汤剂为衡量单味中药配方颗粒是否与其相对应的单味中药饮片临床汤剂基本一致的物质基准;中药配方颗粒生产工艺研究应以标准汤剂为对照,以出膏率、主要成分含量转移率、指纹图谱或特征图谱的一致性为考察指标,对原料、中间体及成品制备过程中的量质传递和物料平衡进行全面研究[2]。该局2018年5月《关于发布古代经典名方中药复方制剂简化注册审批管理规定的公告》规定:经典名方制剂的研制分“经典名方物质基准”研制与制剂研制2个阶段;对汤剂而言,该物质基准又可称为“标准汤剂”或“标准煎液”[3]。经典名方物质基准研究应关注制备过程中受热等因素对质量的影响、关键质量属性的量值传递等[4]。因此,阐明从饮片到标准汤剂的量值传递规律可以为配方颗粒和经典名方制剂工艺研究和质量评价奠定基础。
大黄是一味常用中药,被收载于19个国家的药典[5],大黄所含成分包括蒽醌、蒽酮、鞣质、二苯乙烯、苯丁酮类等[6]。赵曼佳等[7]收集了10批大黄饮片,采用加热回流法制备了标准汤剂,测定了5个游离蒽醌的含量并计算了转移率,建立了标准汤剂UPLC指纹图谱,标定共有峰9个,采用四级杆飞行时间高分辨质谱(quadrupole time-of-flight mass spectrometry,Q-TOF-MS/MS)技术对共有峰进行了鉴定。但该研究未达到标准汤剂“不少于15批”及“不得使用连续回流提取设备”的要求[2],标定的指纹图谱共有峰及测定的成分也偏少。
鉴于此,本实验在前期建立35批大黄饮片HPLC指纹图谱,采用Q-TOF-MS/MS技术鉴定指纹图谱共有峰,并测定芦荟大黄素-8-O-葡萄糖苷(AE8G)、大黄酸-8-O-葡萄糖苷(R8G)、大黄素-1-O-葡萄糖苷(E1G)、大黄酚-1-O-葡萄糖苷(C1G)、大黄酚-8-O-葡萄糖苷(C8G)、芦荟大黄素-3-羟甲基-O-葡萄糖苷(AE3G)、大黄素-8-O-葡萄糖苷(E8G)、大黄素甲醚-8-O-葡萄糖苷(P8G)8个结合型蒽醌和芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚5个游离型蒽醌含量的基础上[6],选择其中23批大黄饮片制备了标准汤剂,建立了标准汤剂HPLC指纹图谱,标定了共有峰,采用Q-TOFMS/MS对共有峰进行了鉴定,测定了标准汤剂中8个结合型蒽醌(AE8G、R8G、E1G、C1G、C8G、AE3G、E8G、P8G)、5个游离型蒽醌(芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚)及饮片和标准汤剂中4个鞣质类成分(没食子酸、儿茶素、表儿茶素、表儿茶素没食子酸酯)、1个二苯乙烯类成分[白藜芦醇4′-O-β-D-(6″-O-没食子酰)-葡萄糖苷(RGG)]的含量,进行了标准汤剂量值传递规律研究。
1 仪器与材料
Waters Alliance高效液相色谱系统,包括e2695分离单元、2998PAD检测器和Empower 3色谱工作站,美国Waters公司;UFLC-DAD-Triple-Q-TOF/MS系统,包括日本岛津公司Shimadzu Prominence UFLC液相色谱仪和美国AB SCIEX公司AB Sciex Triple TOF 4600质谱仪,该质谱仪包括Q-TOF-MS/MS检测器和PeakView1.6质谱分析软件;BT 25S型电子天平,德国Sartorius公司;PB-10型pH计,德国Sartorius公司;SK5200H超声波清洗器,上海科导超声仪器有限公司。
对照品大黄素(批号110751-201512,质量分数98.7%)、芦荟大黄素(批号110795-201710,质量分数98.3%)、大黄素甲醚(批号110758-201616,质量分数99.0%)、大黄酸(批号110757-201607,质量分数99.3%)、大黄酚(批号110796-201621,质量分数99.2%)购自中国食品药品检定研究院;AE8G(批号190115)、R8G(批号180726)、E1G(批号180726)、C1G(批号1180725)、C8G(批号180726)、AE3G(批号190114)、E8G(批号180727)、P8G(批号180730)、没食子酸(批号180114)、儿茶素(批号141224)、表儿茶素没食子酸酯(批号140923)、番泻苷A(批号170508)、番泻苷B(批号CHB170509),质量分数均≥98%,购自成都克洛玛生物科技有限公司;白藜芦醇4′-O-葡萄糖苷(批号38963-95-0)、RGG(批号928340-97-0),质量分数均为98%,购自上海一林生物科技有限公司。
大黄饮片YP1~YP23同文献中S1~S23[6],其中YP1和YP23直接购自中药饮片生产企业,其余均购自国内大中型中医院,YP10~YP17为小包装,其余均为普通包装,饮片来源具有一定代表性,具体信息见表1,经质量检验,均符合《中国药典》2020年版一部大黄项下要求。

表1 大黄饮片信息Table 1 Information of decoction pieces of RRR
2 方法与结果
2.1 大黄标准汤剂及供试品、对照品溶液制备
2.1.1 大黄标准汤剂制备 称取大黄饮片100 g,置烧杯中,加7倍量纯化水浸泡30 min,煮沸,保持微沸30 min,倾出药液,4层纱布滤过;药渣加6倍量纯化水煮沸,保持微沸20 min,倾出药液,4层纱布滤过;2次煎液合并,50 ℃下真空减压浓缩至500 mL,与表1中饮片编号对应,标准汤剂编号TJ1~TJ23。
2.1.2 标准汤剂供试品溶液制备 精密量取“2.1.1”项下制备的标准汤剂1 mL置于50 mL量瓶中,加50%甲醇48 mL,超声处理30 min(功率200 W,频率53 kHz),放冷后加50%甲醇至量瓶刻度,摇匀,滤过,取续滤液,即得。
2.1.3 饮片供试品溶液制备 取大黄饮片粉末(过四号筛)约0.15 g,精密称定,置于25 mL量瓶中,加甲醇24 mL,其余操作同“2.1.2”项。
2.1.4 成分鉴定用混合对照品溶液制备 取所有20个对照品适量,精密称定,加入50%甲醇溶解,混匀,配制成质量浓度为0.73~17.60 μg/mL混合对照品溶液。
2.1.5 含量测定用8种结合型蒽醌混合对照品溶液制备 精密称取8个结合型蒽醌类成分对照品适量,加70%甲醇分别配制成质量浓度为12.12~96.40 μg/mL的含量测定用8种结合型蒽醌混合对照品溶液,取该混合对照品溶液分别稀释100、20、10、2倍,配制成系列质量浓度混合对照品溶液。
2.1.6 含量测定用5种游离型蒽醌类成分混合对照品溶液制备 精密称取5个游离型蒽醌类成分对照品适量,加甲醇分别配制成质量浓度为23.2~104.0 μg/mL的含量测定用5个游离型蒽醌类成分混合对照品溶液,其余操作同“2.1.5”项。
2.1.7 含量测定用5种非蒽醌类成分混合对照品溶液制备 精密称取对照品没食子酸、儿茶素、表儿茶素、表儿茶素没食子酸酯、RGG适量,分别加甲醇溶解并定容,配制成质量浓度分别为1.232、1.870、0.509、0.832、2.300 mg/mL的5种非蒽醌类成分对照品储备液;分别精密量取一定体积的5种非蒽醌类成分对照品储备液,混匀,加甲醇定容,配制成以上5种对照品质量浓度分别为61.60、374.00、76.35、83.20、115.00 μg/mL的含量测定用5种非蒽醌类成分混合对照品溶液,其余操作同“2.1.5”项。
2.2 指纹图谱建立及量值传递分析
2.2.1 色谱条件[6,8]Symmetry C18(250 mm×4.6 mm,5 μm)色谱柱;以甲醇-0.1%磷酸水溶液梯度洗脱:0~10 min,5%~30%甲醇;10~40 min,30%~60%甲醇;40~60 min,60%甲醇;60~70 min,60%~100%甲醇;70~80 min,100%甲醇;柱温30 ℃;体积流量1.0 mL/min;检测波长280 nm。
2.2.2 质谱条件[6,8]流动相为甲醇-0.1%甲酸水溶液,梯度条件同“2.2.1”项。DuoSpray离子源,ESI电离方式,负离子模式检测。离子源喷射电压−4500 V,离子源温度600 ℃,气帘气(CUR)241.317 kPa(35 psi),雾化气(Gas 1)413.685 kPa(60 psi),加热气(Gas 2)413.685 kPa(60 psi)。采用TOFMSIDA-10MS/MS信息采集方式获取质谱信息,参数设置如下:一级质谱解簇电压(DP)−80 V,碰撞能量(CE)−10 eV,TOF-MS累计时间250 ms,母离子扫描范围m/z115~1500;二级质谱子离子质谱扫描范围m/z50~1500,碰撞能量(CE)−35 eV,碰撞能量扩展(CES)15 eV。
2.2.3 指纹图谱测定及对照指纹图谱建立 取YP1~YP23、TJ1~TJ23供试品溶液测定,进样量均为30 μL,采用AIA格式将指纹图谱依次导入《中药色谱指纹图谱相似度评价系统软件(2012版)》,以YP1和TJ1指纹图谱分别作为参照谱,在使用中位数自动匹配的基础上进行多点校正,生成大黄饮片及标准汤剂对照指纹图谱,饮片指纹图谱标定共有峰45个,其中除29、32、37、38和41号峰外的40个传递到了标准汤剂,共有峰个数传递率为88.89%。大黄饮片和大黄标准汤剂指纹图谱及对照指纹图谱见图1、2。

图1 大黄饮片 (YP1~YP23) 指纹图谱及对照指纹图谱 (YPR)Fig.1 Fingerprints of decoction pieces of RRR (YP1—YP23) and reference fingerprint (YPR)
2.2.4 指纹图谱相似度评价 采用《中药色谱指纹图谱相似度评价系统软件(2012版)》分别对大黄饮片和标准汤剂指纹图谱进行相似度计算。结果显示,23批饮片指纹图谱中YP10、YP3、YP16相似度较低,分别为0.458、0.675、0.784,对应的标准汤剂指纹图谱TJ10、TJ3、TJ16相似度也较低,分别为0.584、0.762、0.835,其余批次饮片和标准汤剂指纹图谱相似度均较高。提示标准汤剂的制备工艺稳定、可行,饮片质量较好地传递到了标准汤剂。见表2。
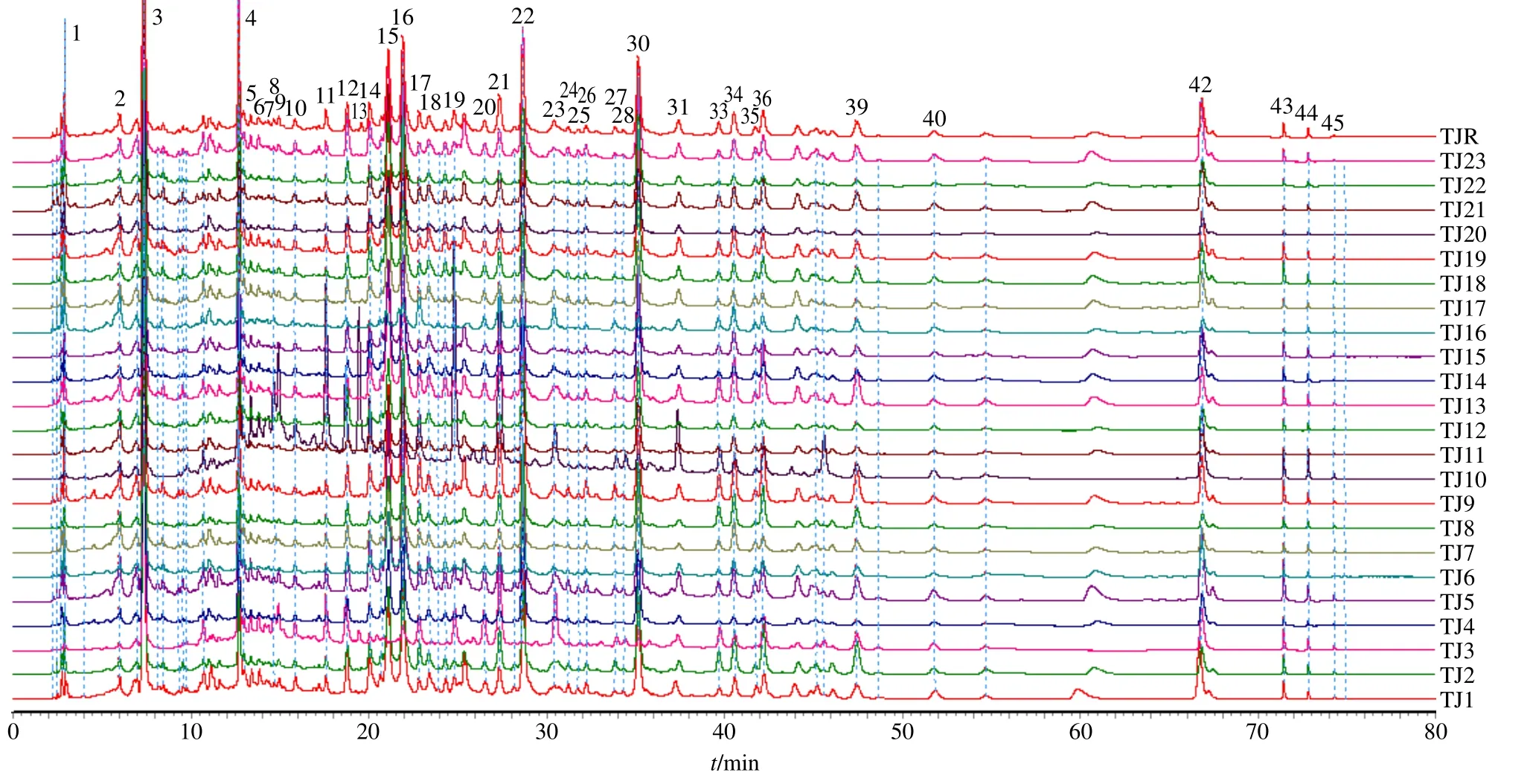
图2 大黄标准汤剂 (TJ1~TJ23) 指纹图谱及对照指纹图谱 (TJR)Fig.2 Fingerprints of standard decoction of RRR (TJ1—TJ23) and reference fingerprint (TJR)
2.2.5 共有峰鉴定 取TJ1供试品溶液采用“2.2.2”项下质谱条件测定,进样量为20 μL,PeakView1.6质谱分析软件提取标准汤剂总离子流图(图3),根据准分子离子 [M-H]−信息判断并得到的一级质谱精确相对分子质量,并根据产生的二级质谱碎片离子信息,通过与前期对照品及饮片测定数据[6]比对,对标准汤剂共有峰进行成分鉴定,结果见表3。结果显示,标准汤剂指纹图谱色谱峰质谱测定数据与前期测定饮片及对照品相应保留时间色谱峰数据基本一致[6],表明饮片指纹图谱传递到标准汤剂指纹图谱的共有峰为同一成分,其中8个结合蒽醌(共有峰17、21、27、33~36、39)、5个游离型蒽醌(共有峰40、42~45)、2个蒽酮类成分(共有峰20、23)、5个鞣质类成分(共有峰3、4、7、10、12)及2个二苯乙烯类成分(共有峰11、19)共计22个成分采用对照品比对确认,其余28个共有峰的鉴定依据见本课题组前期发表的论文[6,9]。
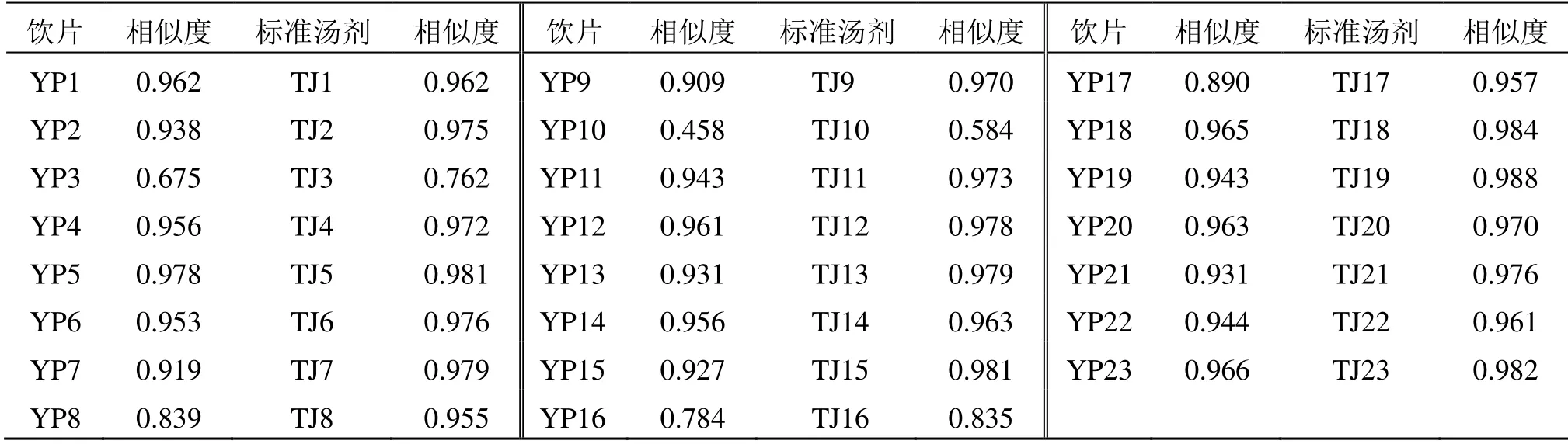
表2 大黄饮片和标准汤剂指纹图谱相似度Table 2 Similarity of fingerprints of decoction pieces of RRR and its standard decoction

图3 大黄标准汤剂总离子流图 (负离子模式)Fig.3 TIC of standard decoction of RRR (negative ion mode)
2.3 蒽醌类成分含量测定及转移率计算
2.3.1 色谱条件 测定波长430 nm,其余同“2.2.1”项。
2.3.2 样品含量测定及转移率计算 提取“2.2.3”项下所得TJ1~TJ23的430 nm色谱图,记录8个结合蒽醌和5个游离蒽醌的峰面积,代入前期建立的回归方程[6]计算标准汤剂中各成分的含量。根据前期饮片含量测定结果计算13个成分的转移率。
转移率=wv/(WvM)[1]
w表示标准汤剂中成分的质量浓度,v表示标准汤剂体积,W表示标准汤剂生药质量浓度,M表示饮片中成分的质量分数
标准汤剂中13个蒽醌类成分含量测定结果见表4,转移率计算结果见表5。
2.4 非蒽醌类成分含量测定及转移率计算
2.4.1 色谱条件 同“2.2.1”项。
2.4.2 专属性考察 取YP1和TJ1供试品溶液及混合对照品溶液进样测定,提取色谱图。结果显示,样品色谱图中5个待测成分基本能达到基线分离,见图4。
2.4.3 线性关系考察 取5种非蒽醌系列质量浓度混合对照品溶液各10 μL,混合对照品溶液10、20、30、40 μL,进样测定,记录6个成分的峰面积,以峰面积(Y)对进样量(X)进行回归处理,得5个成分的回归方程、线性范围及相关系数:没食子酸回归方程Y=2 410 982X-36 296,r=0.999 9,线性范围6.16~2464 ng;儿茶素回归方程Y=732 349X-68 689,r=0.999 9,线性范围37.4~14 960 ng;表儿茶素回归方程Y=734 494X-4435,r=0.999 7,线性范围7.635~3054 ng;表儿茶素没食子酸酯回归方程Y=1 596 107X-24 613,r=0.999 9,线性范围8.32~3328 ng;RGG回归方程Y=3 137 732X-85 754,r=0.999 9,线性范围11.5~4600 ng。
2.4.4 准确度(加样回收率)考察 精密称定S1粉末6份,每份约0.075 g,分别精密添加“2.1.7”项下配制的5个对照品储备液适量(各成分的添加量约等于样品中根据回归方程的计算量),加甲醇24 mL,按“2.1.3”项下方法制备供试品溶液,根据样品含量测定方法测定,计算5个成分的加样回收率及其RSD。结果为没食子酸平均回收率95.05%,RSD为1.89%;儿茶素平均回收率102.99%,RSD为3.64%;表儿茶素平均回收率97.60%,RSD为4.89%;表儿茶素没食子酸酯平均回收率97.65%,RSD为3.74%;RGG平均回收率96.51%,RSD为1.14%,符合《中国药典》分析方法验证指导原则[10]相关要求。
2.4.5 重复性考察 精密称定S1粉末6份,每份约0.15 g,分别按“2.1.3”项下方法制备成6份供试品溶液,根据样品含量测定方法测定,计算6份供试品溶液各成分的质量分数及其RSD。结果没食子酸平均质量分数0.803 mg/g,RSD为1.147%;儿茶素平均质量分数3.717 mg/g,RSD为2.24%;表儿茶素平均质量分数0.188 mg/g,RSD为1.88%;表儿茶素没食子酸酯平均质量分数1.191 mg/g,RSD为2.87%;RGG平均质量分数0.648 mg/g,RSD为2.43%,符合《中国药典》分析方法验证指导原则[10]相关要求。

表3 大黄标准汤剂指纹图谱共有峰Q-TOF/MS鉴定结果Table 3 Identification of common peaks in standard decoction of RRR fingerprint by Q-TOF/MS

表4 大黄标准汤剂中13个蒽醌类成分定量测定结果Table 4 Determination of 13 anthraquinones in standard decoction of RRR

表5 大黄标准汤剂13个蒽醌类成分转移率Table 5 Components transfer rate of 13 anthraquinones of standard decoction of RRR

续表5

图4 大黄饮片 (A)、标准汤剂 (B) 及混合对照品 (C) 的HPLC图Fig.4 HPLC of decoction pieces of RRR (A),standard decoction of RRR (B) and mixed reference substances (C)
2.4.6 耐用性(稳定性)考察 取YP1供试品溶液,分别于制备后0、6、12、18、24、36 h根据样品含量测定方法测定,计算6次测定的各成分峰面积的RSD。以上5种成分的RSD分别为2.75%、2.08%、1.00%、1.41%、3.03%。
2.4.7 样品含量测定及转移率计算 提取“2.2.3”项下所得YP1~YP23和TJ1~TJ23的色谱图,记录5个成分的峰面积,代入回归方程计算饮片和标准汤剂中各成分的含量。同“2.3.2”项下方法计算5个成分的转移率。大黄饮片中5个非蒽醌类成分含量测定结果见表6,标准汤剂中5个非蒽醌类成分含量测定结果见表7,转移率计算结果见表8。

表6 大黄饮片中5个非蒽醌类成分定量测定结果Table 6 Determination of five non-anthraquinone components in decoction pieces of RRR

表7 大黄标准汤剂中5个非蒽醌类成分定量测定结果Table 7 Determination of five non-anthraquinone components in standard decoction of RRR
2.5 标准汤剂pH、出膏率测定及分析
按说明书校准pH计,测定TJ1~TJ23的pH值,每份样品重复3次,取平均值。量取23批大黄标准汤剂各50 mL,置已恒定质量的蒸发皿中,水浴锅上蒸干,烘箱内105 ℃干燥至质量恒定,取出,干燥器内冷却后称定质量,计算出膏率(干浸膏质量/制备50 mL标准汤剂的饮片质量)。每批标准汤剂重复3次试验。结果显示,23批标准汤剂pH在4.84~5.43,均值为5.21,高于赵曼佳等[7]报道的4.15。大黄标准汤剂的出膏率在14.31%~28.63%,均值为23.26%,较文献报道的28.2%[8]略低。出膏率允许的范围为均值加减3倍标准偏差或均值的70%~130%[2],计算发现,23批大黄标准汤剂的出膏率均在均值±3标准差范围内,但有2批超出了均值的70%~130%。结果见表8。

表8 大黄标准汤剂5个非蒽醌类成分转移率及出膏率Table 8 Components transfer rate and extraction ratio of five non-anthraquinone components of standard decoction of RRR
3 讨论
3.1 关于方法学考察
指纹图方法学考察主要包括精密度试验(同一供试品连续进样5次以上,考察色谱峰的相对保留时间和峰面积比值的一致性,采用HPLC法峰面积比值的相对标准偏差RSD不得大于3%)和重现性试验(同一批号供试品5份以上,考察内容同精密度试验,采用HPLC法峰面积比值的相对标准偏差RSD不得大于3%)[11]。本研究指纹图谱方法学考察及结果见前期发表论文[6],精密度试验峰面积比值的RSD为2.92%,重现性试验峰面积比值的RSD为2.98%,均符合技术要求。
根据《中国药典》四部中的通则9101(分析方法验证指导原则)[10],含量测定方法学考察项目包括专属性、准确度、精密度、线性、耐用性。专属性方面,采用色谱法要求附代表性图谱;准确度一般用回收率表示,样品中待测成分含量范围对应有回收率限度要求;精密度主要以重复性体现,样品中待测成分含量范围对应有重复性RSD要求;线性系指在设计的范围内,线性试验结果与试样中被测物浓度直接呈比例关系的能力;耐用性系指在测定条件有小的变动时,测定结果不受影响的承受程度,典型的变动因素有被测溶液的稳定性、样品的提取次数、时间等,液相色谱法中典型的变动因素有流动相的组成和pH值、不同品牌或不同批号的同类型色谱柱、柱温、流速等。
尽管考察项目表述不符合药典要求,13个蒽醌类成分含量测定的方法学考察参见前期发表论文[6],且结果基本符合药典要求,本实验色谱图体现专属性,回收率试验体现准确度,重复性试验体现精密度,线性关系考察体现线性,稳定性试验部分体现耐用性。本研究基本按药典要求对5个非蒽醌类成分含量测定进行了方法学考察,其中耐用性中被测溶液稳定性考察方法及结果见“2.4.6”项,流动相的组成、柱温、体积流量等耐用性试验及结果见本课题组前期建立的大黄药材中非蒽醌类成分含量测定方法[12]。
3.2 关于提取溶剂
前期实验发现,大黄结合型蒽醌并不像大部分文献报道的可溶于甲醇等有机溶剂中,70%甲醇为其合适的溶剂[13],故8个结合型蒽醌对照品溶液的制备采用70%甲醇。赵曼佳等[7]采用离心取上清液的方法制备大黄标准汤剂供试品溶液,可能会导致本来混悬于标准汤剂中的成分流失,测出的成分含量与临床用药实际不符。预实验中比较了50%、70%、100%甲醇制备标准汤剂供试品溶液的效果,发现加入70%和100%甲醇会产生沉淀造成成分损失,加入50%甲醇则溶液基本澄清。
3.3 关于纳入量值规律研究的成分
大黄中所含的各类化学成分具有不同的药理活性[12,14-15],单一组分无法对大黄及其制剂进行全面质量评价[16]。本研究采用Q-TOF-MS/MS对大黄标准汤剂指纹图40个共有峰进行了鉴定,其中8个结合蒽醌、5个游离型蒽醌、2个蒽酮类成分、5个鞣质类成分及2个二苯乙烯类成分共计22个成分采用对照品比对确认,但是由图1和图2可见,蒽酮类成分番泻苷B和番泻苷A、鞣质类成分儿茶素、二苯乙烯类成分白藜芦醇4′-O-葡萄糖苷共计4个成分色谱峰分离度较差,不符合含量测定专属性要求,其余18个成分均建立了含量测定方法,纳入量值规律研究,特别是在同类研究纳入游离型蒽醌基础上,增加了结合型蒽醌。大黄中蒽醌类成分分结合型和游离型2种,其中大部分为结合型蒽醌,但由于其不稳定导致提取纯化比较困难,目前研究较少[9],但近年来大黄结合型蒽醌的作用越来越受到人们的关注[9,17],仅测定游离蒽醌不能全面反映大黄及其制剂的质量[18]。
3.4 关于检测波长
查阅文献发现,多以280、430 nm作为大黄的检测波长,以280 nm检出的色谱峰较多[19],结合蒽醌和游离蒽醌在254、430 nm附近都有吸收峰,但430 nm除蒽醌类成分外的其他成分无吸收[20]。研究过程中提取254、280、430 nm色谱图进行了比较,发现280 nm波长下检出的色谱峰最为全面,因此选择其作为指纹图谱的检测波长。
8个结合型蒽醌中AE8G、R8G、C1G和C8G分别在254、410 nm附近有最大吸收,E1G、E8G、P8G分别在280、420 nm附近有最大吸收,AE3G在254、430 nm附近有最大吸收[9];5个游离型蒽醌中芦荟大黄素、大黄酸、大黄酚分别在254、430 nm附近有最大吸收,大黄素、大黄素甲醚分别在280、440 nm附近有最大吸收[17]。提取410、430 nm色谱图比较发现,两者并无较大差异,为避免其他成分的干扰,故选择430 nm作为蒽醌类成分含量测定波长。提取5个待测非蒽醌类成分色谱峰的紫外吸收光谱图发现,5个成分均在280 nm附近有最大吸收,所以选择280 nm为非蒽醌类成分含量测定波长。
3.5 关于成分转移率
23批大黄标准汤剂5个游离型蒽醌合计的转移率为5.44%~37.95%,均值为11.03%,赵曼佳等[7]报道的10批大黄标准汤剂的相应数据分别为11.16%~28.14%和19.87%,两者结果差异有待分析。但总体来讲,游离型蒽醌的平均转移率偏低,其主要原因可能是游离型蒽醌苷元为脂溶性成分,在水煎液中难以溶出,已有多篇文献提到标准汤剂中脂溶性成分转移率低的问题[21]。大黄素甲醚、大黄酚和大黄素的平均转移率分别只有1.41%、1.90%和3.66%,芦荟大黄素为13.70%,大黄酸则相对偏高,为37.31%,可能是由于5个成分在加热煎煮过程中结构发生了相互转化而导致大黄酸的含量比例提高[7,22-25],计算结果显示,23批大黄饮片中大黄酸含量的平均值占5个游离型蒽醌合计平均值的比例为24.24%,标准汤剂中相应数据为81.97%,与文献报道的药材中大黄酸占游离蒽醌的比例为17.39%,而提取物中该比例提高到37.71%[22]。8个结合型蒽醌转移率相对较高,均值在35.96%~81.74%,8个成分合计平均转移率为44.83%,尽管这些结合型蒽醌在加热煎煮和浓缩过程也有部分转化成了水不溶性的游离型蒽醌[13],但测定这些水溶性成分来计算转移率更为合理[26]。
大黄中鞣质类成分含量高达10%~30%[27],本次标定的大黄标准汤剂指纹图谱40个共有峰中17个为鞣质类成分,大黄鞣质分水解型和缩合型2类,单体分别为没食子酸和儿茶素[28],酚酸和多元醇通过苷键或酯键形成可水解鞣质[29]。本实验测定的大黄标准汤剂中的4个鞣质类成分中表儿茶素和没食子酸平均转移率分别高达303.18%和233.00%,表儿茶素没食子酸酯最低,为58.71%,其原因可能为含苷键和酯键的鞣质类成分易催化水解[29],如有研究表明,在加热过程中表儿茶素没食子酸酯易水解为表儿茶素和没食子酸[30],这样就导致标准汤剂中表儿茶素没食子酸酯转移率偏低,表儿茶素和没食子酸相对含量提高。同样,其他结构中含没食子和表儿茶素结构的以酯键和苷键链接的缩合物也可能水解生成表儿茶素和没食子酸。
利益冲突所有作者均声明不存在利益冲突
