Negative thermal expansion in NbF3 and NbOF2:A comparative theoretical study*
2021-05-24MingyueZhang张明月ChunyanWang王春艳YinuoZhang张一诺QilongGao高其龙andYuJia贾瑜
Mingyue Zhang(张明月), Chunyan Wang(王春艳),2, Yinuo Zhang(张一诺),Qilong Gao(高其龙),†, and Yu Jia(贾瑜)2,,‡
1International Laboratory for Quantum Functional Materials of Henan,and School of Physics and Microelectronics,Zhengzhou University,Zhengzhou 450052,China
2Key Laboratory of Special Functional Materials of Ministry of Education,and School of Materials Science and Engineering,Henan University,Henan 475001,China
Keywords: negative thermal expansion,fluorides,lattice dynamics calculation,average atomic volume,negative Gr¨uneisen parameters
1. Introduction
Negative thermal expansion (NTE) is an interesting physical phenomenon, in which volume shrinks rather than expands with increasing temperature.[1–3]The presence of NTE materials offers a promising possibility to control the thermal expansion in optical, electronic, and engineering applications.[4]So far, a number of families of NTE materials have been identified such as alloy,[5]oxides,[6–9]fluorides,[10,11]cyanides,[12,13]Prussian blue analogues (PBAs),[14,15]metal-organic frameworks,[16,17]manganese nitrogen compounds,[18,19]and so on. However,compared with the number of positive thermal expansion (PTE)materials, the quantity and variety of NTE materials are very small, thus limiting their further applications. Hence, nowadays, exploring or predicting new NTE materials and further understanding NTE mechanism are always urgent and challenging.
Due to the presence of the unique ionic bond character,the NTE behavior has been found in some ReO3-type metal fluorides. For example, the simplest cubic structure of ScF3with the wide-temperature-window (10–1100 K) and large isotropic NTE (60–110 K, αl≈−14 ppm·K−1) has been reported by experiment in 2010. It is the only one found in single ReO3-type metal fluorides. The double-ReO3-type, such as CaZrF6and CaHfF6were found with much larger isotropic NTE (αl≈−18 ppm·K−1and −20 ppm·K−1at 100 K) than that of ScF3.[11]Then based on the chemical general formula AIIBIVF6, several NTE materials were found, such as CaNbF6,[20]MnZrF6,[21]and CaTiF6.[22]However, the NTE fluoride materials have also very litter. For the thermal expansion control,it always adopts the chemical modification in cations. For example, chemical substitution in (Sc1−xMx)F3(M =Al, Ti, Fe and Ga)[22–25]or by redox intercalation in Lix(Sc0.9Fe0.1)F3,[26]the crossover from NTE to zero thermal expansion(ZTE)and finally to PTE was widely observed.It also has been observed that it could been adjusted by the F-excess such as TiZrF7−x[7]and Mg1−xZr1+xF6+2x.[28]Recently,we notice that the NbF3has the cubic ReO3type crystal structure, which represents a typical structure of TMO3−xFx(TM=Nb,Ta,Ti,Mo,and W).[29,30]Similar thermal expansion control behavior occurs in many oxides materials such as NH3in ZrW2O8[31]and Zr0.5Hf0.5VPO7.[32]More recently,Li et al.[33]reported that the chemical bond had larger effect on the NTE behavior in MCo(CN)6(M=Fe, Ti, Co). It naturally leads us to assume that if we adjust part chemical bond in NbF3,the NTE behavior maybe have some change. It is an interesting idea to tailor negative NTE by designing the chemical bond.
In this work,the NTE,electronic structure,lattice dynamics of NbF3and NbOF2are investigated by first-principles calculations with density functional theory and the quasiharmonic approximation. We show that the NTE of NbOF2is smaller than that of NbF3and the driving force of NTE in NbOF2comes from the transverse vibration of the F and O atoms, while the vibration amplitude of the O atoms is much smaller than that of the F atoms.
2. Computational details
All the results are calculated using density functional theory (DFT) as implemented in the Vienna ab into simulation package (VASP) code.[34]The ion–electron interaction is depicted by projector augmented wave (PAW) method[35]and the exchange and correlation effects are described by the generalized gradient approximation with PBE functional.[36]The energy cutoff is 520 eV and an 11×11×11 k-points mesh has been used to obtain the total energy convergence. The convergence criteria for the total energy and ionic forces are set to 10−8eV and 10−3eV/˚A,respectively.
Phonon disoersion spectra of NbF3and NbOF2are calculated by frozen phonon method within a 2×2×2 supercell and post processed by PHONOPY code.[37]A 2×2×2 grid is used for the Brillouin-zone integration. The thermodynamic properties are simulated through the quasi-harmonic approximation(QHA)theory. In this approximation,the volume dependent phonon frequencies at finite temperatures are introduced as a part of the anharmonic effect. In our previous works,this method provided reasonable and interesting description of the dynamic properties of materials below the melting point.[38–41]Within the QHA, the phonon contribution to the Helmholtz free energy F(V,T)can be written as

where E(V) is the total static energy form DFT calculations,Fvib(V,T)stands for the vibration energy and is calculated by
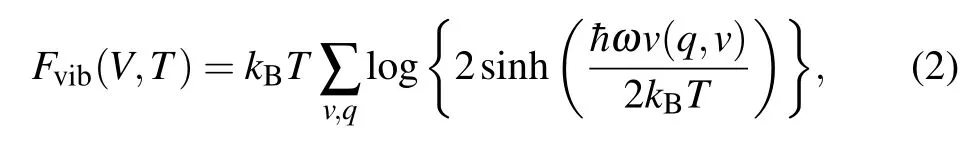
where ωv(q,v) is the phonon frequency at a fixed volume V and a given vector q. In order to obtain the effect of temperature on volume, 11 diffferent volumes are selected near the optimized equilibrium position.
3. Results and discussion
3.1. Crystal structures and coefficient of thermal expansion
As mentioned in the introduction,NbF3and NbOF2crystallize in cubic structure with the space group (Pm-3m), and the unit cell contains one formula unit with 4 atoms.[29,42]Nb and O(F) atoms lie at 1a(0, 0, 0) and 3d(0.5, 0, 0) Wyckoff sites, respectively. The crystal structure is composed of NbF6or NbO6octahedra,which is isostructural with ScF3,as shown in Figs. 1(a) and 1(b). In our calculation, the lattice constants of NbF3and NbOF2are 4.11860 ˚A and 4.10951 ˚A,respectively. The temperature dependence of the cell volume and the coefficients of thermal expansion(CTE)of NbF3and NbOF2are present in Figs. 1(c) and 1(d), respectively. It is found that the cell volumes at a finite temperature for both NbF3and NbOF2are smaller than those at 0 K, exhibiting a typical NTE behavior. The largest CTE for NbF3is about−31×10−6K−1at 100 K,while for NbOF2,the largest CTE is about −9×10−6K−1. Hence, this implies that the NTE behavior could be tailored by the introduction of O atom in NbF3. More importantly, the underlying mechanism of such large differences between them in NTE should be noticed and need to study.

Fig.1. (a),(b)Geometric structures of NbF3 and NbOF3,(c),(d)temperature dependence of the cell volume and CNTE of NbF3 and NbOF2,respectively.
3.2. Negative thermal expansion mechanism and role of O atoms in NbOF2
In order to investigate the underlying mechanism responsible for the NTE of NbF3and the role of O atoms in NbOF2,we have calculated the corresponding phonon spectrum, partial phonon density of states(DOS),and Gr¨uneisen parameters of NbF3and NbOF2compounds, as shown in Figs. 2(a) and 2(b). It is shown that the phonon dispersion curves along the high-symmetry lines of the Brillouin zone(BZ)have no imaginary frequencies,indicating that NbF3and NbOF2are dynamically stable. In the region of high frequency,it is the vibration of F atoms for NbF3while for NbOF2,it is the vibration of O atoms. In the region of low frequency, compared with NbF3,the substitution of O makes the frequencies at M and R points increase,and the frequency at M point is higher than that at R point. As shown in DOS(Figs. 2(a)and 2(b)right), the contribution to vibrational frequency below 200 cm−1is mainly from F atoms.However,the vibration of F atoms is suppressed in NbOF2, which suggests that the vibrations associated with F atoms are easy to be excited predominantly at low temperatures for NbF3, while it is inhibited in NbOF2. Figures 2(c)and 2(d) display the calculated Gr¨uneisen parameters as a function of vibrational frequency in Brillouin zone(inset along high symmetry directions). In general,the Gr¨uneisen parameter reflects the anharmonic effect,i.e.,the nonlinear vibrational modes of atoms.[43]If the sum of negative Gr¨uneisen parameters(γi)for the excited modes is larger than that of the positive ones,the CTE of the material will be negative.[43]Similar to ScF3, obviously, the M(0.5 0.5 0) and R(0.5 0.5 0.5)points at Brillouin-zone boundary have the largest negative values.[44]The corresponding negative Gr¨uneisen parameters of NbOF2are much smaller than those of NbF3,thus introducing a smaller CTE.

Fig.2. (a),(b)Calculated phonon dispersion curves and partial density of states(DOS).(c),(d)Calculated Gr¨uneisen parameters as a function of vibrational frequency(inset along high symmetry directions)for NbF3 and NbOF2,respectively.

Fig.3. Atomic displacement schemes for low frequency vibrational modes at M and R points. (a),(c)NbF3 and(b),(d)NbOF2.
Since the NTE is closely related to the modes with relative large negative Gr¨uneisen parameters, we have therefore studied these vibrations modes to further shed light on the mechanism. The low-energy vibrational modes of NbF3and NbOF2at M and R points are shown in Fig. 3. At M point,for NbF3, F atoms have transverse vibrations in the ab-plane and the metal Nb atoms are stock-still, resulting in a librational motion of undistorted NbF6octahedra. The coupling rotations of the adjacent octahedra shorten the effective length of neighboring metal–metal atoms, therefore contributing to the NTE behavior. For the NbOF2, as well as NbF3, it is the transverse vibrations of F atoms in the ab-plane that contribute to the NTE.At R points,for NbF3,it is the vibration mode of triple merger along three sides. For NbOF3,it is not the same three vibration modes,so the merger is broken up.It should be noted that at the metal–O(or F)–metal linkage,the amplitude of vibration of O atoms is smaller than that of F atom.
On the other hand, the strength of chemical bonding has great impact on the NTE behavior of framework compounds.[33]To further illustrate the relationship between chemical bonding and NTE phenomenon,the charge densities on(010)plane are shown in Figs.4(a)and 4(b)for NbF3and NbOF2, respectively. We can see an obvious ionic bonding character between Nb and F,while in NbOF2,an evident covalent character between Nb and O,and an ionic bonding character between Nb and F. Hence, the bond of Nb–O is much stiffer than the Nb–F bond, so the transverse vibration of O atoms is more difficult than that of F, and hence the NbOF2system exhibits a small NTE behavior.

Fig.4. Charge densities of NbF3 (a)and NbOF2 (b)on(010)plane.

Fig.5. (a)Energy difference versus non-metal atoms displace. (b)The anisotropic displacement ellipsoids at 300 K,the atoms are represented as ellipsoids that reflect the distribution of thermal displacements. The distributions of displacements of the anions are strongly elongated in the transverse directions perpendicular and parallel to the bond for(c)NbF3 and(d)NbOF2,respectively.
3.3. Average atomic volume and atomic displacement parameters
It should be noted that the NbF3and NbOF2have an open-framework structure. The NTE behavior of openframework compounds has strong relationship with the structure flexibility.[2,15]Recently,we noted the concept of average atomic volume (AAV) to explore new NTE materials.[45]Interesting, the AAV of NbF3is about 18 ˚A3at 300 K, which is much larger than the critical AAV (~16 ˚A3). While the AAV of NbOF2is about 16.6 ˚A3,which is also larger the critical AAV but samller than that of NbF3. Hence,the reduction of AAV should be a key factor for the difference of NTE behavior by the introduction of O atoms. The anisotropic atomic displacement parameters(ADPs)also could quantify the effect of the induction of O atoms in NbF3and NbOF2. Like most of open-framework NTE compounds, the NTE comes from the transverse thermal vibration of bridge atoms.[10,12,46]Obviously,the transverse vibration of F atoms in NbF3and NbOF2is almost the same,while larger than that of O atoms(Fig.2).Hence,the introduction of O atoms weakens the transverse vibration of bridge atoms, thus producing the inhibiting effect.The energy barrier versus non-metal atoms displace is investigated to reflect the transverse vibration potential of bridge atoms.[47]The energy potential curves of F atoms in NbF3and both F and O atoms in NbOF2are obtained by changing the atom position along the vibrating direction, as shown in Fig.5(a).
Obviously,the energy difference in NbOF2is steeper than that in NbF3, which indicate that the corresponding atomic vibration in NbOF2is more difficult than that in NbF3. In all, considering that there is no obvious difference of lattice constants between NbF3and NbOF2from our calculation,we conclude that,if we introduce the O atom to NbF3,the CTE of NTE in NbOxF3−xcan be easily tuned from −9×10−6K−1to −30×10−6K−1without the structures distorted. This provides an ideal NTE material for realizing composite material with zero thermal expansion.
4. Conclusion
In summary, the NTE behavior and mechanism of cubic NbF3and NbOF2are investigated by using first-principles calculations within density functional theory and the quasiharmonic approximation. NbF3shows strong NTE due to the transverse vibration modes of F atoms, once the 1/3 F atoms are substituted by O atoms, the transverse vibration of the F atoms is hindered, thus the NTE will be reduced. Combined with the eigenvectors of phonon modes corresponding to the NTE behavior, electronic properties, and total-energy of atoms-displaced configurations,the NTE of NbF3is larger than that of NbOF2due to the produce of covalent chemical bond by O atoms. The present work not only predicts two new NTE compounds,but also provides an insight on thermal expansion control.
Acknowledgment
Zhang Mingyue thanks Dr. Li Chong for his technical support.
杂志排行

Chinese Physics B的其它文章
- Corrosion behavior of high-level waste container materials Ti and Ti–Pd alloy under long-term gamma irradiation in Beishan groundwater*
- Degradation of β-Ga2O3 Schottky barrier diode under swift heavy ion irradiation*
- Influence of temperature and alloying elements on the threshold displacement energies in concentrated Ni–Fe–Cr alloys*
- Cathodic shift of onset potential on TiO2 nanorod arrays with significantly enhanced visible light photoactivity via nitrogen/cobalt co-implantation*
- Review on ionization and quenching mechanisms of Trichel pulse*
- Thermally induced band hybridization in bilayer-bilayer MoS2/WS2 heterostructure∗