SiO2包覆对Ni2P/Al2O3催化剂结构和萘加氢性能的影响
2021-05-21荆洁颖杨志奋王九占刘道诚李文英
荆洁颖,李 泽,杨志奋,王九占,刘道诚,李文英
(太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室,山西 太原 030024)
煤焦油作为煤热解过程中的副产物,年产量在2 000万t以上[1-2],其中低温煤焦油性质与原油相近,但其组成中含有大量的芳烃(主要以双环和三环化合物为主)[3],与喷气燃料中的碳数分布在同一范围(C8~C15)。因此,可通过催化加氢将低温煤焦油中的芳烃转化为氢化芳烃或环烷烃,作为喷气燃料的理想组分提升其热安定性。然而,煤焦油与石油原料组成差异巨大,传统石油化工加氢催化剂难以满足煤焦油制喷气燃料的需求,因此,如何获得高性能加氢催化剂是低温煤焦油制高性能喷气燃料的关键。
目前在芳烃加氢方面,商用的催化剂以过渡金属硫化物为主,虽然其具有一定的抗中毒能力,但其加氢活性较低[4-6];贵金属催化剂表现出最佳的加氢饱和能力,但其成本高,且活性位点易被含S、含N等杂原子化合物所毒化[7];过渡金属碳化物、氮化物和磷化物在芳烃加氢反应中都表现出很高的加氢活性,因此被广为研究[8-9]。其中,过渡金属磷化物催化剂兼具高的加氢活性和抗中毒性能[10-13],在芳烃加氢领域具有很大潜力。在所有的磷化物催化剂中,Ni2P催化剂因其具有特殊的晶体形貌和电子结构表现出很高的本征加氢活性[14-15],有望成为新一代高效芳烃加氢催化剂。但由于非负载型Ni2P催化剂存在比表面积小、活性相分散度、机械强度及散热性不佳的问题,影响其反应活性。因此,研究者大多借助载体的高比表面积分散活性组分,进而暴露更多的活性位点,提高催化剂的活性。
就负载型Ni2P催化剂而言,不同的载体对Ni2P的生成、分散度和催化剂的活性都有着重要影响。目前使用的载体主要有γ-Al2O3、活性炭、SiO2以及分子筛等[16-19]。为提高催化剂活性,很多学者就载体改性进行了研究。笔者课题组前期在Al2O3中引入Ce进行掺杂改性,使得Al2O3中四配位的Al数量大大减少,从而抑制了AlPO4的生成,提高了磷源的利用率,促进了Ni2P在Al2O3上的生成,催化剂的萘加氢活性也大幅提高[20-21]。LI等[22]考察了ZrO2掺杂改性的Al2O3载体负载Ni2P活性组分的加氢脱硫和加氢脱氮活性,研究结果显示,当ZrO2掺杂量为20%时催化剂的活性最高。此外,OYAMA等[14]研究发现,小晶粒的Ni2P可暴露出更多的Ni(2)位,产生更多的加氢活性位点,从而使得催化剂的加氢活性得以提高。
为充分发挥次磷酸盐歧化法制备小晶粒Ni2P的优势,基于笔者课题组前期采用次磷酸盐歧化法制备Ni2P/Al2O3催化剂的相关研究结果[23-24],为进一步促进Ni2P在载体表面的分散并生成更多的Ni2P物种,笔者将通过对Al2O3表面进行SiO2包覆的方法改变前驱体中镍物种、催化剂中Ni2P与载体之间的相互作用,使得在适宜的载体与金属相互作用下生成更多Ni2P物种并实现更高的分散,从而提高催化剂的芳烃加氢性能。
1 实验部分
1.1 SiO2-Al2O3载体的制备
采用化学液相沉积法[25]制备表面包覆SiO2的Al2O3载体,制备的具体步骤如下:将1 g干燥后的粉末状γ-Al2O3加入30 mL环己烷中搅拌5 min,然后向上述溶液中加入一定量的正硅酸乙酯并搅拌5 h,然后静置至溶液出现分层现象(上层为透明液体、下层为白色沉淀);此时用吸管移除上层液体,将剩余白色沉淀在60 ℃水浴加热4 h,所得白色粉末置于马弗炉中以3 ℃/min的升温速率升温至500 ℃并恒温4 h进行焙烧,得到不同SiO2包覆量的Al2O3载体。当正硅酸乙酯用量为0,0.2,0.4,0.6,0.8和1.0 mL时,相应的SiO2包覆量为0,5.3%,10.7%,16.0%,21.4%和26.7%,将所得载体记做Al2O3-xSi,xSi表示Al2O3外层包覆SiO2的质量分数为x%。
1.2 Ni2P/SiO2-Al2O3催化剂的制备
以不同SiO2包覆量的Al2O3-xSi为载体,采用次磷酸盐歧化法制备Ni2P/SiO2-Al2O3催化剂,其中催化剂中活性组分Ni2P的负载量为10%。具体步骤为:按照Ni,P物质的量比为0.5称取计算量的乙酸镍(Ni(CH3COO)2·4H2O)和次磷酸铵(NH4H2PO2)溶于一定量去离子水中,在搅拌状态下混合均匀得到澄清的绿色溶液,向上述溶液中加入0.42 g Al2O3-xSi载体,并在室温下持续搅拌8 h,将所得物质放置于60 ℃ 干燥12 h。干燥后的固体粉末放入管式炉内进行还原。管式炉的升温程序为:从室温开始以2 ℃/min 升至400 ℃,恒温2 h,催化剂的还原过程始终在H2气氛下进行,H2流速设为80 mL/min。为避免还原后的催化剂在空气中再次被氧化,还原后的催化剂需在60 mL/min 1%(体积分数)O2,N2气氛下钝化2 h,所得催化剂记做Cat-xSi催化剂,xSi表示Al2O3外层包覆SiO2的质量分数为x%。
1.3 Ni2P/SiO2-Al2O3催化剂结构表征
采用Agilent 720ES电感耦合等离子体原子发射光谱仪(ICP-OES)测定催化剂中镍和磷的含量。其操作过程如下:将36 mg催化剂放入消解罐中,加4 mL硝酸、1 mL氢氟酸和1 mL双氧水,然后将消解管放入消解仪开始微波消解。消解完成后的样品经除酸后定容稀释进行ICP-OES测试。
催化剂的晶相结构采用日本理学的Rigaku Ultima Ⅳ型X射线衍射粉末衍射仪进行分析,测试的管电流和管电压分别为40 mA和40 kV,扫描速度为4(°)/min,扫描范围为10°~80°。
催化剂的还原性能在美国Micromeritics公司的Autochem II 2920型化学吸附仪上进行测试。H2-TPR测试过程为:称取0.05 g催化剂前驱体放入U型管中,先通Ar在120 ℃下预处理2 h(Ar流量为40 mL/min),待U型管温度降至50 ℃,再切换到10%(体积分数)H2/Ar以10 ℃/min的速率升温至800 ℃进行程序升温实验(H2/Ar流量为50 mL/min),由热导检测器(TCD)记录实验信号。
催化剂的比表面积和孔结构分析由美国康塔公司的Quantachrome Autosorb-iQ获得。分析前,样品在240 ℃,0.5×10-4MPa进行3 h的预处理,以除去水分和气体杂质,然后在-196 ℃下进行N2吸脱附实验。采用BET法计算催化剂的比表面积,采用BJH模型计算孔径分布。
催化剂的表面元素状态由日本岛津公司的Kratos Analytical AXIS Supra光电子能谱仪进行测试。表面形貌及其粒径分布由美国赛默飞世尔科技公司的Talos F200X 型透射电子显微镜进行测试,加速电压为200 kV。
催化剂的酸性由美国麦克默瑞提克公司的Autochem II 2920型化学吸附仪表征得到。具体操作步骤如下:称取0.05 g的催化剂放入U型管中,先通10%(体积分数)H2/Ar对催化剂表面的氧化层进行还原(H2/Ar流量为50 mL/min,升温程序为20 ℃/min升温到400 ℃,恒温2 h),还原完成后切换气路为He,待U型管冷却至100 ℃以后再切换气路为NH3/He,催化剂吸附NH3时间为30 min。吸附完成后再次切换气路为He对装置进行吹扫,并开启升温程序(从100 ℃以10 ℃/min的速率升温至500 ℃),吸附于催化剂表面的NH3开始程序升温脱附,同时由TCD记录实验信号。
采用CO脉冲实验间接对催化剂的活性位点数量进行表征。首先取0.1 g催化剂置于U型管中,通10%(体积分数)H2/Ar对催化剂表面的氧化层进行还原(H2/Ar流量为50 mL/min,升温程序为20 ℃/min升温到400 ℃,恒温2 h),还原完成后切换气路为Ar,待U型管冷却至100 ℃以后再切换气路为5%(体积分数)的CO/Ar进行脉冲实验,定量环容量为0.1 mL,直到吸附饱和。
1.4 催化剂的活性评价
采用连续固定床反应器[24]对催化剂活性进行评价。采用内径为8 mm,长度为460 mm的不锈钢管为反应器,催化剂装填量为6.4 mg(粒径0.25~0.42 mm)。反应前,催化剂在温度400 ℃、压力1 MPa下,采用100 mL/min H2还原2 h。还原完毕后,调节反应条件为:温度340 ℃,H2压力为4 MPa,氢油比为600,重时空速(WHSV)为20.8 h-1,最后以6 mL/h的速率通入3%(质量分数)萘/四氢萘的正癸烷溶液,反应开始计时,产物取样间隔时间为1 h。反应后产物组成采用GC-2010型气相色谱(日本岛津)进行分析。采用内标法对产物进行定量分析,以十二烷为内标物,乙醇为溶液。各个催化剂的活性评价实验平均做3次,在每次萘/四氢萘加氢反应中,反应时间为8 h;每间隔1 h取得的加氢产物与内标物混合后进行取样分析,平行测试3次,每次反应计算的相对偏差若在3%以内,则认为实验数据可靠。转化率按下式计算:

(1)
式中,X为萘或四氢萘的转化率;Nin为反应器入口萘或四氢萘的摩尔量;Nout为反应器出口萘或四氢萘的摩尔量。
2 结果与讨论
2.1 Cat-xSi催化剂微观性质表征
2.1.1NiO/Al2O3-xSi催化剂的还原性
为确定SiO2是否成功包覆于γ-Al2O3表面,首先对比了SiO2,γ-Al2O3以及Al2O3-16.0Si三种不同载体负载NiO催化剂的还原性能,结果如图1所示。可以看到:NiO/SiO2在500 ℃和680 ℃附近可观察到2个明显的H2消耗峰,其中500 ℃附近的H2消耗峰归属于SiO2表面游离态NiO物种的还原,而680 ℃附近的H2消耗峰则归属于和SiO2相互作用较强的NiO物种的还原,在焙烧过程中这部分Ni物种与SiO2结合紧密,导致其还原温度较高;NiO/γ-Al2O3的H2消耗峰出现在660 ℃附近,但其峰宽较大(520~800 ℃),这是由于催化剂表面的NiO物种与γ-Al2O3的相互作用强度不同所致;NiO/Al2O3-16.0Si在500 ℃和700 ℃附近可观察到2个H2消耗峰,对比NiO/SiO2和NiO/Al2O3的H2消耗峰可以发现,NiO/Al2O3-16.0Si在500 ℃附近的H2消耗峰归属于γ-Al2O3表面包覆的SiO2上NiO物种的还原,而700 ℃的H2消耗峰则归属于γ-Al2O3上的NiO物种的还原。由此可见,通过化学液相沉积法可实现将SiO2包覆在γ-Al2O3表面。此外,对比不同载体负载的NiO的H2消耗峰的峰面积大小可知,NiO物种在SiO2上的还原度最高,Al2O3-16.0Si次之,在γ-Al2O3上的还原度最低,这也充分说明了金属镍与不同载体之间的相互作用不同。其他Al2O3-xSi载体负载NiO的H2-TPR图同NiO/Al2O3-16.0Si的规律一致。

图1 不同载体负载NiO的H2-TPR图Fig.1 H2-TPR profiles of NiO load on different supports
2.1.2Cat-xSi催化剂的元素组成
采用ICP-OES测得的不同SiO2包覆量Cat-xSi催化剂中镍和磷的含量见表1。由表1中数据可知,包覆SiO2后,Cat-xSi催化剂中Ni的质量分数在7.6%~8.3%,与Ni的理论质量分数十分接近,表明次磷酸盐歧化法可保证Ni按照添加量成功负载于Al2O3-xSi载体上。而P的实际质量分数较其理论值均有所减少,这是由于次磷酸盐歧化后生成的PH3未完全参与Ni2P的生成,存在部分流失所致[16],因此实际的Ni,P物质的量之比较理论值略高。

表1 Cat-xSi催化剂的元素组成Table 1 Element composition of Cat-xSi catalysts
2.1.3Cat-xSi催化剂的晶相结构
不同SiO2包覆量Cat-xSi催化剂的XRD谱图如图2所示。由图2可知,所有催化剂的XRD谱图均十分相似,仅在2θ=31.85°,37.54°,39.28°,60.55°以及66.60°处出现了γ-Al2O3的特征峰,均未观察到Ni2P的特征峰,推测是因为制得的Ni2P颗粒在催化剂表面高度分散(图3),因此未被XRD检测到。D’AQUINO等制备的Ni2P/Al2O3催化剂在其XRD谱图中亦未能观察到Ni2P特征峰,笔者认为是因为Ni2P颗粒在催化剂表面的高度分散导致XRD未能检测到其存在[26]。此外,SiO2的包覆并未引起Cat-xSi催化剂的XRD衍射峰的峰强度和半峰宽出现明显差别,且随着SiO2包覆量的增加亦未观察到变化,可见SiO2在γ-Al2O3表面的包覆对于Ni2P的晶相结构影响不大。

图2 Cat-xSi催化剂的XRD谱图Fig.2 XRD patterns of Cat-xSi catalysts
2.1.4Cat-xSi催化剂的比表面积和孔结构
不同SiO2包覆量Al2O3-xSi载体及其负载Ni2P后的Cat-xSi催化剂的比表面积和结构参数见表2和3。可以看到:Al2O3-xSi载体的比表面积、总孔容均随SiO2包覆量的增加而减小,表明SiO2成功负载在γ-Al2O3上;而Al2O3-xSi载体的孔径却呈先增大后减小的趋势,这可能是因为在载体的制备过程中,少量的SiO2会先将γ-Al2O3的微孔堵塞,表现出平均孔径有所增加,随着SiO2包覆量的增加,一些介孔也被堵塞,故表现出平均孔径减小。

表2 不同载体的比表面积和孔结构Table 2 BET specific surface area and pore structure of different supports

表3 不同催化剂的比表面积和孔结构Table 3 BET specific surface area and pore structure of Cat-xSi catalysts
负载Ni2P后的Cat-xSi催化剂与其对应的载体相比,比表面积、总孔容和平均孔径均有不同程度的减小,其中未包覆SiO2的Cat-0Si催化剂在这些参数方面的减少更加显著,而Cat-xSi催化剂较其对应的载体在这些参数方面的减少程度要小,这可能是由于Al2O3载体在包覆了SiO2之后,载体与Ni物种之间的相互作用减弱,使得浸渍在催化剂表面的Ni物种更容易被还原,这与H2-TPR的结果一致;此外γ-Al2O3载体的平均孔径比Al2O3-xSi载体小,因此在催化剂制备过程中沉积的磷盐更易堵塞Ni2P/Al2O3的孔道,使得Cat-xSi催化剂表现出较Cat-0Si较大的比表面积、总孔容和平均孔径。
2.1.5Cat-xSi催化剂的表面形貌和粒径分布
采用透射电子显微镜(TEM)观测不同SiO2包覆量Cat-xSi催化剂的形貌和粒径分布情况,如图3所示。首先,通过对高分辨TEM照片中条纹间距的测量确定了Ni2P颗粒的形成;其次,可以看出以Al2O3-xSi作载体制备的Cat-xSi催化剂,其表面的Ni2P颗粒较Cat-0Si要分散地更均匀,对应的Ni2P粒径也更小。WU等[25]用SiO2包覆γ-Al2O3负载Ni颗粒之后也发现了Ni颗粒粒径变小的现象。随着γ-Al2O3表面的SiO2包覆量增加,催化剂中的Ni2P颗粒呈现先减小后增大的趋势,当SiO2包覆量为16.0%(质量分数)时,Cat-16.0Si催化剂中Ni2P粒径最小,为4 nm;表明适量SiO2包覆可促进Ni2P的分散,生成更多小粒径的Ni2P颗粒。
2.1.6Cat-xSi催化剂的表面酸性
采用NH3-TPD对不同SiO2包覆量Cat-xSi催化剂的表面酸性进行了分析(图4)。据报道,小于230 ℃的NH3脱附峰归属于弱酸位点的脱附,230~500 ℃的NH3脱附峰归属于中强酸位点的脱附,而大于500 ℃的NH3脱附峰则归属于强酸位点的脱附[27-28]。由图4可知,未包覆SiO2的Cat-0Si催化剂可在220 ℃和590 ℃左右观察到2个NH3脱附峰,且590 ℃左右NH3脱附峰较220 ℃附近的NH3脱附峰面积更大,表明Cat-0Si催化剂表面存在弱酸位点和强酸位点,而且强酸位点的数量更多。与Cat-0Si催化剂对比可以发现,所有包覆SiO2的Cat-xSi催化剂在220 ℃左右的NH3脱附峰较590 ℃的NH3脱附峰更明显,这可能是由于SiO2的引入覆盖了γ-Al2O3的强酸中心,而与此同时SiO2又和γ-Al2O3形成了Si-O-Al键,使得表面的弱酸位点增加。进一步分析Cat-xSi催化剂的NH3脱附峰可以发现,尽管SiO2的包覆量不同,但是所有催化剂在590 ℃左右的NH3脱附峰的面积十分接近,而在220 ℃附近的NH3脱附峰却随着SiO2包覆量的增加而增大,由此可以推断,包覆在γ-Al2O3表面的SiO2会优先落到其强酸位点,然后随着SiO2引入量的增加,多余的SiO2再与γ-Al2O3形成Si—O—Al键,从而表现出催化剂表面弱酸性位点增多,这一变化规律同WU[25]和梁旭[29]等的报道一致。

图4 Cat-xSi催化剂的NH3-TPD图Fig.4 NH3-TPD profiles of Cat-xSi catalysts
采用Py-IR对载体及不同SiO2包覆量Cat-xSi催化剂表面酸性位点数量以及不同酸中心的分布状况进行了监测,表4列出了不同催化剂表面的L酸中心和B酸中心的数量。可以看到:包覆SiO2后的Al2O3-16.0Si载体表面B酸中心数量较γ-Al2O3表面略增,而L酸中心数量较γ-Al2O3有明显的减少。负载Ni2P之后的Cat-0Si和Cat-16.0Si催化剂与γ-Al2O3和Al2O3-16.0Si相比,其B酸和L酸的酸量均有不同程度的下降,显然活性组分的加入覆盖了部分酸性位点。但是值得注意的是,Cat-0Si催化剂表面的B酸量较γ-Al2O3减少了2.3 μmol/g,而Cat-16.0Si表面的B酸量较Al2O3-16.0Si仅减少了0.6 μmol/g,推测不同催化剂表面的B酸位点数量较其载体的B酸量减少程度不同可能是由于催化剂表面不同的磷盐沉积量产生了不同数量的P—OH这种B酸位点所致。
2.1.7Cat-xSi催化剂的表面元素电子性质
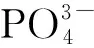

表4 催化剂表面不同的酸性中心分布Table 4 Distribution of different acid center on surface of catalyst

图5 Cat-xSi催化剂中Ni 2p和P 2p的XPS谱Fig.5 XPS spectra of Ni 2p and P 2p on Cat-xSi catalysts
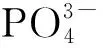

表5 Cat-xSi催化剂中Ni 2p和P 2p的结合能和Ni,P物质的量之比Table 5 Binding energy of Ni 2p and P 2p on Cat-xSi catalysts and the molar ratio of Ni and P
2.2 Cat-xSi催化剂的活性评价
2.2.1SiO2包覆的影响
以低温煤焦油中典型芳烃物质—萘作为模型化合物进行催化剂性能评价,结果如图6(a)所示。活性评价条件为:空速20.8 h-1,反应温度340 ℃,氢气压力4 MPa,氢油比600,进料量6 mL/h,催化剂用量6.4 mg,采用CO脉冲吸附描述催化剂中活性位数量[29]。

图6 Cat-xSi催化剂的萘和四氢萘加氢转化率Fig.6 Conversion of naphthalene and tetralin on Cat-xSi catalysts
由图可知,Cat-0Si催化剂在8 h内的萘转化率保持在56%左右,而所有包覆SiO2的Cat-xSi催化剂萘转化率较Cat-0Si均有明显的提高,且Cat-16.0Si催化剂的萘加氢活性最高,为72%。由H2-TPR、NH3-TPD和XPS的结果可知,SiO2的引入覆盖了Al2O3表面的强酸位点,削弱了Ni物种和P物种与载体之间的相互作用,使得催化剂表面的Ni物种和P物种更容易还原,Cat-xSi催化剂中生成了较Cat-0Si催化剂更多的Ni2P物种,在催化剂表面暴露出更多的活性位点;并且从TEM照片和粒径分布图中可知,SiO2的包覆促进了载体表面Ni物种的分散,形成了颗粒更小的Ni2P,因此暴露出更多加氢活性位点Ni(2)位;此外,从Py-IR的数据可知,SiO2包覆在Al2O3表面后,表面形成了Si—O—Al键,催化剂表面的B酸位点数量略有增加,从而使芳烃更易被活化。因此,SiO2包覆的Cat-xSi催化剂表现出较未包覆SiO2的Cat-0Si催化剂更高的芳烃加氢活性。当SiO2包覆量为16.0%(质量分数)时,Cat-16.0Si催化剂中的Ni2P颗粒最小(仅4 nm)、表面暴露的Ni2P数量最多、活性位的数量最多(CO吸附量为26.8 μmol/g)且表面的B酸位点数量最多(2.8 μmol/g),因此Cat-16.0Si催化剂表现出最高的芳烃加氢活性。
但值得注意的是,在所有催化剂的萘加氢产物中均只有四氢萘,而未检测到十氢萘。基于这一现象作出以下推测:一方面可能是在较高空速(20.8 h-1)下,反应物在催化剂表面的停留时间太短,并且在反应体系中存在的萘分子较其首环加氢产物四氢萘更易在催化剂表面吸附,因此四氢萘还未继续加氢就已发生脱附;另一方面可能是由于四氢萘的加氢比萘加氢更为困难,而Cat-xSi催化剂的加氢能力有限,仅能使萘加氢到四氢萘。
为明确上述现象的具体原因,以四氢萘为原料,分别采用Cat-0Si和Cat-16.0Si催化剂在同样的条件下进行了四氢萘加氢活性评价,评价结果如图6(b)所示。可以看到:2种催化剂的四氢萘加氢产物均只有十氢萘,未检测到任何裂解产物。在8 h内,Cat-16.0Si和Cat-0Si催化剂的四氢萘转化率都比较稳定,且Cat-16.0Si催化剂的四氢萘加氢转化率(38%)高于Cat-0Si催化剂(21%)。但是比较同一催化剂的萘加氢转化率和四氢萘加氢转化率可知,Cat-0Si和Cat-16.0Si的四氢萘加氢转化率较其萘加氢转化率都要低很多。由此可见,Cat-xSi催化剂具有使四氢萘加氢到十氢萘的能力,但四氢萘加氢要比萘加氢更困难;此外,在萘加氢反应的产物仅有四氢萘的原因可归结于在较高的重时空速(WHSV=20.8 h-1)下,四氢萘分子在催化剂表面的停留时间较短以及四氢萘在与萘的竞争吸附中处于劣势,导致四氢萘还未参与加氢就已经从催化剂表面脱附。
2.2.2反应前后Cat-16.0Si催化剂的结构变化
为了进一步探究Cat-16.0Si催化剂具有高芳烃加氢活性的原因,对反应前后Cat-16.0Si催化剂的晶相结构进行了分析(图7)。由图7可知,反应前后催化剂的晶相结构没有明显变化,且均只能观察到Al2O3的特征峰,未能检测到Ni2P特征峰的出现。推测反应过程中Cat-16.0Si催化剂中Ni2P比较稳定,未发生颗粒的长大。通过对比反应前后Cat-16.0Si催化剂的TEM照片和Ni2P粒径分布图(图8),可以看出:反应前后催化剂中的Ni2P颗粒始终保持小颗粒,并未出现明显的长大,这一结论与反应后Cat-16.0Si催化剂的XRD结果一致。

图7 反应前后Cat-16.0Si催化剂的XRD谱Fig.7 XRD patterns of fresh and spent Cat-16.0Si catalysts

图8 反应前后Cat-16.0Si催化剂形貌及其Ni2P粒径分布Fig.8 Morphologies and Ni2P particle size distribution of fresh and spent Cat-16.0Si catalysts
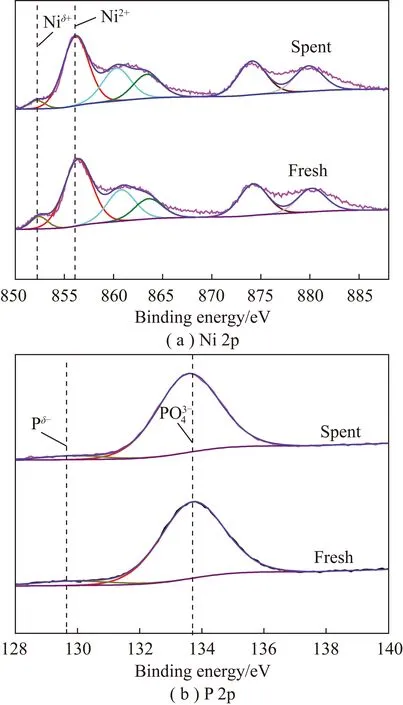
图9 反应前后Cat-16.0Si催化剂中Ni 2p和P 2p的XPS谱Fig.9 XPS spectra of Ni 2p and P 2p on fresh and spent Cat-16.0Si catalysts
3 结 论
(1)SiO2的包覆对Ni2P/Al2O3催化剂的还原性、晶粒粒径、酸性、活性位点数量等均有很大影响,进而引起了Ni2P/Al2O3催化剂的萘加氢反应性能差异。
(2)在反应温度340 ℃、氢气压力4 MPa、空速为20.8 h-1下,所有包覆SiO2的Cat-xSi催化剂均表现出较未包覆SiO2的Cat-0Si催化剂更高的萘加氢活性(包覆SiO2的Cat-xSi催化剂萘转化率均高于65%,未包覆SiO2的Cat-0Si催化剂萘转化率仅56%),其中Cat-16.0Si催化剂的萘加氢活性最高(72%)。
(3)Cat-16.0Si催化剂高的加氢活性归因于Al2O3表面SiO2的包覆,一方面SiO2的存在使得镍物种与载体间的相互作用更为适宜,导致Cat-xSi催化剂中镍物种更容易被磷化为Ni2P,形成了更多小粒径的Ni2P颗粒(Cat-16.0Si的平均粒径为4.0 nm),暴露出更多的活性位点(Cat-16.0Si的CO吸附量为26.8 μmol/g);另一方面适量的SiO2包覆提高了活性组分Ni2P中Niδ+的缺电子程度,引入了B酸位点,促进了芳烃在催化剂中的吸附。
