高效液相色谱法测定棉菜籽中甲萘威残留量
2021-05-18刘晓燕
◎ 刘晓燕
(北京世纪科环生态农业研究院,北京 101102)
甲萘威又名西维因,是一种氨基甲酸酯类杀虫剂,由于杀虫谱广和毒性较低,在农业上应用广泛。目前,甲萘威残留检测多采用HPLC-FLD 柱后衍生法和HPLC-PDA 法。HPLC-FLD 柱后衍生法的灵敏度较高,但前处理复杂,衍生剂稳定性较差,耗时较长,在某种程度上也提高了检测成本,不宜广泛使用。HPLCPDA 法前处理简便,准确性和重复性较好,但在检测油性产品时,加标回收的数据达不到满意的效果。为了克服现有方法的不足,建立起广泛的应用基础,本实验采取与GB/T 5009.21—2003[1]完全不同的前处理方法,样品经乙酸甲醇提取、QuECHERS 试剂净化后,测定甲萘威的含量。
1 材料与方法
1.1 材料和试剂
棉菜籽,购自超市。
乙二胺-N-丙基硅烷化硅胶(PSA)、石墨化碳黑(GCB)、丙酮等为分析纯;甲醇、乙腈为色谱纯;弗罗里硅土柱:500 mg/6 mL;C18 固相萃取柱: 500 mg/6 mL;艾杰尔ASB-C18色谱柱:250 mm×4.6 mm, 5.0 μm。
甲萘威标准品:1 000 μg·mL-1,购自农业部环境保护科研监测所。
1.2 仪器及设备
LC-20AT 高效液相色谱仪(配二极管阵列(PDA)检测器、日本岛津公司)、FA2204B 分析天平(天津天马衡基仪器有限公司)、KDC-160HR 低速离心机(安徽中科中佳科学仪器有限公司)
1.3 标准储备液配制
准确移取浓度为1 000 μg·mL-1的标准物质溶液 1.0 mL,用甲醇定容至10 mL 配制成甲萘威标准储备液100.0 mg·L-1。
1.4 样品的提取与净化
称取2 g(精确至0.01 g)棉菜籽,加入1 g 氯化钠和10 mL 1%乙酸甲醇,涡旋3 min,静置30 min,加入适量的无水MgSO4冷却后涡旋3 min,5 000 r·min-1离心5 min,取5 mL 上层转移至分散性固相萃取管 (750 mg 硫酸镁、150 mg PSA、100 mg GCB)中,充分涡旋,5 000 r·min-1离心5 min,然后取2 mL 上清液转移至玻璃离心管中,40 ℃氮气吹干,20%甲醇定容1 mL,涡旋3 min,0.22 μm 有机滤膜过滤,待测。
1.5 色谱条件
色谱柱为艾杰尔ASB-C18:250 mm×4.6 mm, 5.0 μm;流动相:甲醇+水(80+20,V/V);流速:1.0 mL·min-1;检测波长:280 nm;柱温:40 ℃;进样量:10 μL。
2 结果与分析
2.1 实验条件优化
2.1.1 提取剂的选择
对样品棉菜籽中的甲萘威分别采用了正己烷、乙腈、丙酮、1%乙酸甲醇进行提取。如表1 所示,在相同的添加水平下,用1%乙酸甲醇为提取剂时,回收率高达88.8%,且甲萘威在酸性条件下会更加的稳定,杂质较少,峰形更好,因此本文最终选用1%乙酸甲醇作为提取溶剂。在1%乙酸甲醇为提取溶剂时,不用添加有机溶剂可达到去油效果,且对甲萘威的检测结果无影响。

表1 不同提取溶剂对化合物的回收率表
2.1.2 净化方法选择
GB/T 5009.21—2003[1]中的净化方法非常繁琐,且称样量大。本方法采取1%乙酸甲醇进行提取,通过无水硫酸镁进行有效的脱水后,将得到的提取液分别移取到分散性固相萃取柱、弗罗里硅土柱和C18 固相萃取柱中。平行测定3 次得到试验结果。经试验证实,C18 固相萃取柱和弗罗里硅土柱的平均回收率为65%~70%,分散性固相萃取柱的平均回收率能达到88%~90%,因此本文选用分散性固体萃取柱。
2.1.3 分散性固相萃取柱填料的配比
根据王晓菁等的研究成果[2],加入50 mg PSA 和150 mg 无水硫酸镁进行前处理,净化效果不理想,因此实验中增加了配比量。因填料中添加GCB、PSA 能进行有效的净化,所以填料设计分别为1 组:600 mg无水硫酸镁和100 mgPSA,填料;2 组:900 mg 无水硫酸镁、100 mg PSA 和15 mg GCB;3 组:750 mg 无水硫酸镁、150 mg PSA 和100 mg GCB。添加相同量的甲萘威标准储备液进行加标回收试验,将不同配比的填料回收率进行比较。根据比对选择3 组,其配比为750 mg 无水硫酸镁、150 mg PSA 和100 mg GCB,该配比下的填料净化回收率最高。3 种不同填料配比实验回收率结果见表2。
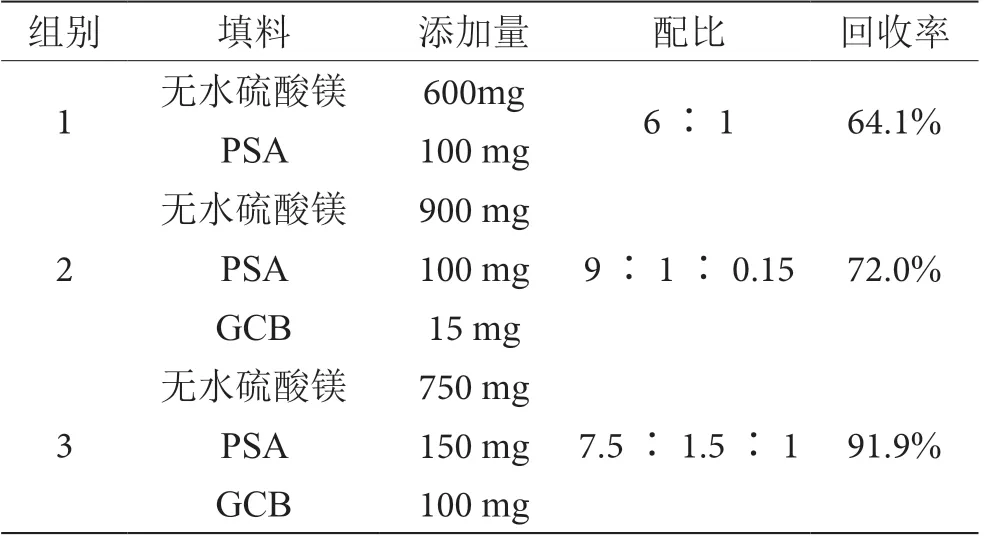
表2 3 种不同填料配比实验结果表
2.1.4 波长的选择
将甲萘威标准溶液在高效液相色谱(带PDA 检测器)仪上200 ~300 nm 的范围内进行扫描测定,确认在280 nm 的波长处有最高吸收峰,且杂质也最少。因此选择检测波长为280 nm。分别以甲醇和水、乙腈和水作为流动相。
2.1.5 流动相的选择
甲醇和乙腈作为流动相时分离度均可以达到甲萘威检测的基本要求。考虑到乙腈成本比甲醇要高,所以选择甲醇水作为流动相。实验证明当甲醇与水的体积比为80 ∶20 时,甲萘威峰形对称性较好,且能有效的分离杂质,所以将流动相定为甲醇∶水(80 ∶20,V/V)。
2.1.6 色谱柱的选择
色谱柱选用了艾杰尔ASB-C18 型色谱柱和岛津ODS-C18 型色谱柱进行色谱分离甲萘威,实验结果表明,两种色谱柱所分离出的目标化合物的峰形,分离度都满足要求,考虑岛津ODS-C18 型色谱柱比艾杰尔ASB-C18 型色谱柱的成本要高,所以采用艾杰尔ASB-C18 型色谱柱(250 mm×4.6 mm,5.0 μm),分离的峰形满意,保留时间为19.910 min。
2.2 线性范围
根据色谱条件对0.10 mg·L-1、1.0 mg·L-1、2.0 mg·L-1、5.0 mg·L-1和10 mg·L-1甲萘威标准工作曲线进行测定,测定结果见表3。以峰面积为纵坐标,浓度为横坐标进行绘制标准曲线,得回归方程y=141 016x-8 918.3,相关系数为0.999 8。

表3 甲萘威标准曲线数据表
2.3 检出限与定量限
在上述样品处理方法及色谱条件下,以信噪比 S/N=3 计,测得甲萘威检出限为0.1 mg·kg-1;以信噪比S/N=10 计,测得甲萘威定量限为0.5 mg·kg-1。根据GB 2763—2019[3]规定油料和油脂甲萘威中的最大残留量为1 mg·kg-1,因此该方法完全满足棉菜籽中甲萘威残留量分析的要求。
2.4 加标回收率的测定
对棉菜籽的阴性样品做加标回收试验,分别称取已处理的样品2.0 g,添加0.500 mg·kg-1、1.000 mg·kg-1、 1.500 mg·kg-1的甲萘威标准工作溶液进行做加标试验,并设置空白样,平行测定6 次。结果见表4,在 0.500 mg·kg-1、1.000 mg·kg-1、1.500 mg·kg-1的3 个 不同的加标水平回收率在82.9%~91.9%,相对标准偏差(RSD)在2.17%~4.25%,说明该方法的准确度能够满足甲萘威的测定要求。

表4 加标回收率实验结果表
2.5 精密度
准确配制甲萘威标准工作溶液1.0 mg·L-1,根据上述色谱条件,进样量10 μL,平行测定7 次。甲萘威色谱峰的保留时间波动小于0.1 min,如表5 所示,峰面积相对标准偏差(RSD)小于10%,表明方法精密度良好。
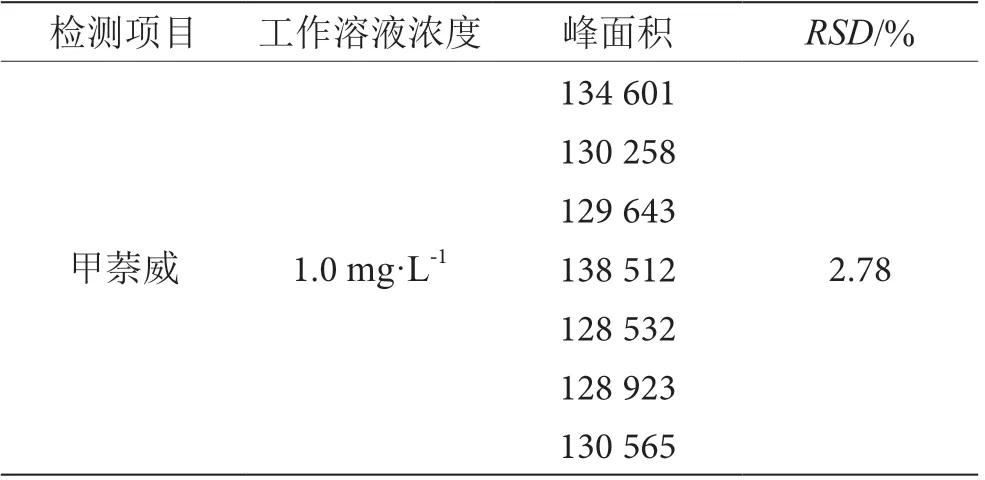
表5 精密度检测结果表
3 结论
本文采用QuECHERS 前处理技术以高效液相色谱法测定棉菜籽中的甲萘威,比GB/T 5009.21—2003 的方法操作简单、准确性高、重现性好,降低了实验中的成本,提高了操作人员的实验效率。GB 2763—2019[3]对甲萘威的最大检出量为0.1 mg·kg-1,用本方法检测出的甲萘威检出限小于0.1 mg·kg-1,定量限小于0.5 mg·kg-1,因此该方法适用于在棉菜籽上的残留量的检测。
