超高效液相色谱-串联质谱法在 食品安全检测中的应用
2021-05-18郑茜玥
◎ 曹 维,谭 杰,郑茜玥
(武汉海关技术中心,湖北 武汉 430050)
国家标准《食品安全国家标准 食品中兽药最大残留限量》(GB 31650—2019)限定了食品动物中甲砜霉素等药物残留限量,但食品中有关氯霉素及甲硝唑等药物残留事件报道仍较多[1]。兽药残留为目前食品安全面临的重大问题之一[2-3]。基于液相色谱-串联质谱法为食品中多种兽药残留快速定性筛查的首选方法,且具有分析效率高、灵敏度高的优势[4],本文就超高效相色谱-串联质谱法在动物源性食品检测中应用效果进行探究。
1 材料与方法
1.1 仪器与试剂
(1)仪器。LC-30 超高效液相色谱仪(日本岛津公司)、ATRAP 5500 三重四极杆液相色谱质谱联用仪(美国AB SCIEX 公司)、Multi Reax 全能型振荡器(德国Heidolph 公司)、Centrifuge 5804 高速离心机(德国Eppendorf 公司)、SCIENTZ-950E 超声波提取器(宁波新芝生物科技公司)、Autovap S60 氮吹仪(美国ATR 公司)、XS-205 电子分析天平(瑞士Mettler-Toledo 公司)、Milli-Q 超纯水制备系统(美国MILLI-PORE 公司)、HYPERSEP Retain-PEP 固相萃取柱(美国赛默飞公司)、陶瓷均质子(美国安捷伦公司)以及0.22 μm PFP 微孔滤膜(上海安谱科学仪器有限公司)。
(2)试剂与样品。氯霉素、氟甲砜霉素、甲砜霉素标准品;猪肉粉中氯霉素、氟苯尼考分析质控样;乙腈、甲醇和甲酸;实验用水。样本为当地农贸市场所采购猪肉、牛肉及羊肉,总计68 批。
1.2 方法
1.2.1 样本处理
样品切碎后,准确称量5 g 样品置于50 mL 聚丙烯具塞离心管中,加50 μL 混合同位素内标溶液,放置30 min 后,加pH=9.0 磷酸盐缓冲液5 mL,涡旋振荡5 min 后,加乙腈5 mL,混匀后,以1 000 r·min-1冷冻离心共10 min,去上清液后置另一个离心管中,加乙酸乙酯10 mL,涡旋30 s 后,静置分层,于1 000 r·min-1冷冻离心10 min,去上层液,置15 mL 聚丙烯离心管,40 ℃以下水浴中,氮气吹至近干,残留物加0.3 mL甲醇溶液,加5.7 mL 磷酸盐缓冲液,混匀待净化。
1.2.2 溶液配制
取标准品氯霉素、氟甲砜霉素、甲砜霉素,溶于甲醇配制为标准储备液,浓度为1 mg·mL-1,置于-20 ℃ 下避光保存,有效期6 个月。量取标准储备液适量,用甲醇稀释至10 μg·mL-1标准工作液,保存在2 ~8 ℃,有效期1 个月。按1.2.1 处理后,获取阴性样品,按照混合标准,配制为质量浓度1 ng·mL-1、5 ng·mL-1、10 ng·mL-1、 20 ng·mL-1、50 ng·mL-1、75 ng·mL-1和100 ng·mL-1混合液。
1.2.3 分析条件
(1)色谱条件。色谱柱:Waters ACQUITYUPLC BEH C18 柱;柱温:40 ℃;样品室温度:10 ℃;流速: 0.4 mL·min-1;流动相A:0.1%甲酸水溶液;流动相B:甲醇;梯度洗脱程序:0 ~3.0 min,90%A →60%A; 3.0 ~3.5 min,60%A →15%A,3.5 ~4.0 min, 15%A →10%A,4.0 ~4.5 min,10%A →90%A, 4.5 ~6.0 min,90%A;进样量:10 μL。
(2)质谱条件。离子源为电喷雾电离源,负离子扫描,多反应模式监测。毛细管电压:2.5 kV(负离子)和4.0 kV(正离子);离子源温度:150 ℃,脱溶剂气温度:500 ℃,脱溶剂气流量:1 000 L·h-1,碰撞气流量:0.13 mL·min-1。氯霉素、甲砜霉素和氟甲砜霉素的离子对、去簇电压、碰撞气能量和碰撞室出口电压见表1。

表1 质谱条件相关参数表
1.2.4 线性分析
阴性样品处理液获取混合配制,1 ng·mL-1、 5 ng·mL-1、20 ng·mL-1、50 ng·mL-1、75 ng·mL-1和 100 ng·mL-1系列基质标准曲线,并上机测定。将目标化物定量离子峰面积归为纵坐标,质量浓度为横坐标,绘制标准曲线。
1.2.5 精密度测定
对质量浓度为10.0 ng·mL-1的3 种目标物标准溶 液,开展6 次重复测定,测定精密度。
1.2.6 样品测定
吸取1 mL 上清液加入使用前依次用3 mL 甲醇、3 mL 水活化处理过的弱阴离子固相萃取柱中,待样品以低于1.0 mL·min-1的流速全部通过固相萃取柱,再分别用3 mL 去离子水和3 mL 甲醇淋洗,流速为 1 mL·min-1,应用10 mL 2%氨水-甲醇溶液洗脱,收集洗脱液在45 ℃水浴中氮吹至近干,应用60%甲醇-水 溶液定容至1 mL,涡旋混匀1 min,上机测定。按照液相色谱-串联质谱条件测定样品和标准工作溶液,样品中待测物质的保留时间与标准溶液中待测物质的保留时间偏差在±2.5%。定量测定时采用标准曲线法测定,定性是应与浓度相当标准工作溶液的相对峰度一致。
2 结果与分析
2.1 质谱条件和色谱条件优化
质谱条件优化:4 种目标化合物响应值具有差异性,配制100 ng·mL-1目标物溶液,流动注射三通进样方式在正离子模式下进行母离子全扫描,设置碰撞电压1 eV,调节锥孔电压使准分子离子峰的响应值最大,当椎体电压为30 V 时氯霉素和氟甲砜霉素产生的准分子离子[M+H]+并分别为m/z192、m/z202,响应值最高。当碰撞电压为18 eV 时,甲砜霉素可产生子离子m/z160。
色谱条件优化:根据试样中被测样液的含量情况,选取待测物的响应值在一定线性相应范围内的浓度进行测定。在1.2.3 色谱条件下氯霉素、氟甲砜霉素、甲砜霉素参考保留时间分别为6.1 min、4.9 min、4.5 min。 各个目标化合物分离良好,且色谱峰峰型良好。
2.2 线性范围
测得目标化合物在1.0 ~100 ng·mL-1呈现良好的线性关系,线性方程为y=6 878.82x,相关系数r ≥0.992 9。
2.3 精密度测定结果
如表2 所示,对质量浓度为10.0 ng·mL-1的3 种目标物标准溶液,开展6 次重复测定,测得结果相对标准偏差为0.4%~2.3%,说明仪器具备良好精密度。
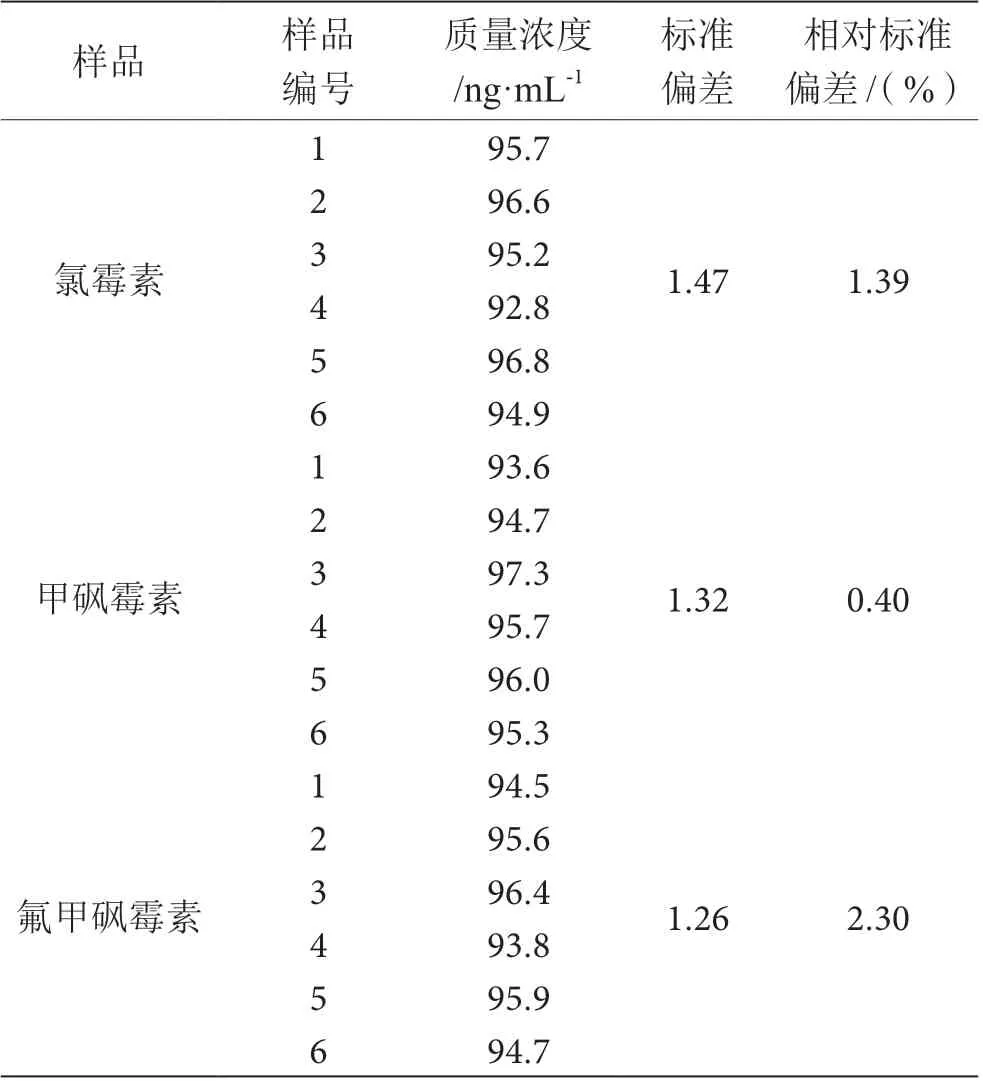
表2 精密度测定结果表
2.4 样品测定结果
如表3 所示,氯霉素、甲砜霉素、氟甲砜霉素测定低限为0.1 μg·kg-1。本次方法中检出值为0.3 μg·kg-1,低于标准方法检测低限、检测限(SN/T 2289—2009),氯霉素、甲砜霉素、氟甲砜霉素测定低限均为0.5 μg·kg-1;表明上述方式灵敏度高。
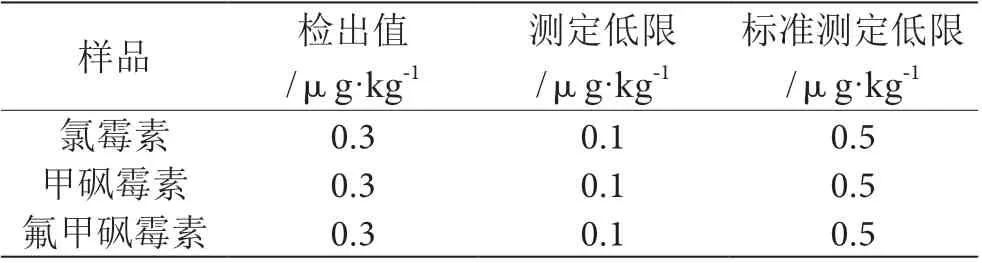
表3 样品测定结果表
3 结论
超高效相色谱-串联质谱法特点为高效、快速,为食品安全及质量检测中开辟新的途径。相较传统高效液相色谱,采取更小微粒柱填充材料及较高泵压,分离效果更为显著[5]。研究结果表明,目标化合物在1.0 ~100 ng·mL-1呈现良好的线性关系,线性方程为y=6 878.82x,相关系数r ≥0.992 9。氯霉素、甲砜霉素、氟甲砜霉素测定低限为0.1 μg·kg-1。精密度测定,相对标准偏差0.4%~2.3%,说明方法具备良好的精密度。本次方法中,氯霉素类药物检出值为0.3 μg·kg-1,低于标准方法检测低限、检测限。综上,超高效相色谱-串 联质谱法简单、快捷、灵敏度高,可满足食品及保健食品市场监管及检验需求。
