吡咯的硝基亚胺衍生物的结构稳定性和爆轰性能研究
2021-05-17赵飞李璐琳
赵飞 李璐琳
摘 要:通过在吡咯环中引入硝基亚胺,本研究设计了一系列吡咯的硝基亚胺衍生物,旨在寻找高能量密度化合物。其间通过计算G3MP2能级的生成热、键离解能和键序,对分子稳定性进行了深入的探讨。结果表明,本研究设计的分子具有足够的热稳定性和动力学稳定性。为评价其作为高能量密度化合物的潜在应用价值,笔者利用Kamlet-Jacobs方程计算了其爆轰速度和爆轰压力。基于计算结果,本文筛选出四种硝基亚胺吡咯衍生物(D1,即1,2,3,4-四硝基亚胺吡咯-1H-吡咯;D2,即2,3,4,5-四硝基亚胺吡咯-1H-吡咯;D3,即1,2,3,5-四硝基亚胺吡咯-1H-吡咯;E,即1,2,3,4,5-五硝基亚胺吡咯-1H-吡咯)作为可能的高能量密度分子,供进一步研究。
关键词:高能量密度材料;热稳定性;Kamlet-Jacobs方程;爆轰性能
中图分类号:TQ560.1文献标识码:A文章编号:1003-5168(2021)03-0116-03
Study on the Structural Stability and Detonation Performance
of Nitroimine Derivatives of Pyrrole
ZHAO Fei LI Lulin
(School of Chemistry and Materials, Guizhou Education University,Guiyang Guizhou 550018)
Abstract: By introducing nitroimine into the pyrrole ring, this study designed a series of pyrrole nitroimine derivatives, aiming to find compounds with high energy density. In the meantime, by calculating the heat of formation of the G3MP2 energy level, the bond dissociation energy and the bond sequence, the molecular stability was discussed in depth. The results showed that the molecules designed in this study had sufficient thermal and dynamic stability. In order to evaluate its potential application value as a high-energy density compound, the author used the Kamlet-Jacobs equation to calculate its detonation velocity and detonation pressure. Based on the calculation results, this paper screened out four nitroimine pyrrole derivatives (D1, namely 1,2,3,4-tetranitroimine pyrrole-1H -pyrrole; D2, namely 2,3,4,5-tetranitroimine pyrrole-1H -pyrrole; D3, namely 1,2,3,5-tetranitroimine pyrrole -1H -pyrrole; E, namely 1,2,3,4,5-pentanitroimine pyrrole-1H -pyrrole) as possible high energy density molecules for further study.
Keywords: high energy density materials;thermal stability;Kamlet-Jacobs equation;detonation performance
近几十年来,寻找高能量密度材料(HEDMs)的工作一直在进行[1],其中以C-H-N-O型化合物最引人注目,人们发现了许多稳定性高、爆轰性能优良的物种[1]。根据这些发现,设计新型高能密度化合物的一般策略是在含氮量较大的母体中引入能量基团。
吡咯属于氮杂环,天然含氮量高于烷烃。硝基亚胺与吡咯环之间的相互作用已被证明能有效地稳定这些衍生物,从理论上证实了吡咯具有优良的爆轰性能。由此可见,吡咯是设计新型高能量密度材料的理想母体。与硝基亚胺相比,硝胺能更有效地改善爆轰性能。两个硝胺基团就能使四唑盐的比重增加到令人惊讶的2.177 g/cm3,其优于六硝基亚胺六氮杂异伍兹烷(CL-20),几乎是已知C-H-N-O型炸药的最高值[2]。这意味着下一代炸药不需要CL-20这样的复杂结构,含有硝胺基团的平面分子是增加比重和爆轰特性的关键。
为此,本文先后用硝基亚胺取代吡咯中的氢原子,设计了新的高能密度化合物,并采用Kamlet-Jacobs方程的密度泛函方法对其衍生物的热稳定性、动力学稳定性和爆轰特性进行了研究。
1 计算方法
在G3MP2水平上,本文采用G03包对所有几何构型进行了优化[3]。其间通过等键反应计算吡咯的硝基亚胺取代衍生物的生成热(HOFs)[4]。本文采用经典Kamlet-Jacobs方程预测化合物的爆速(D)和爆轰压力(P)。化学键的强度可以用键离解能(BDE)来判断,化合物的稳定性也受到鍵的解离能的影响,本文通过设计等键反应来计算解离能。
2 結果和讨论
2.1 生成热和分子稳定性
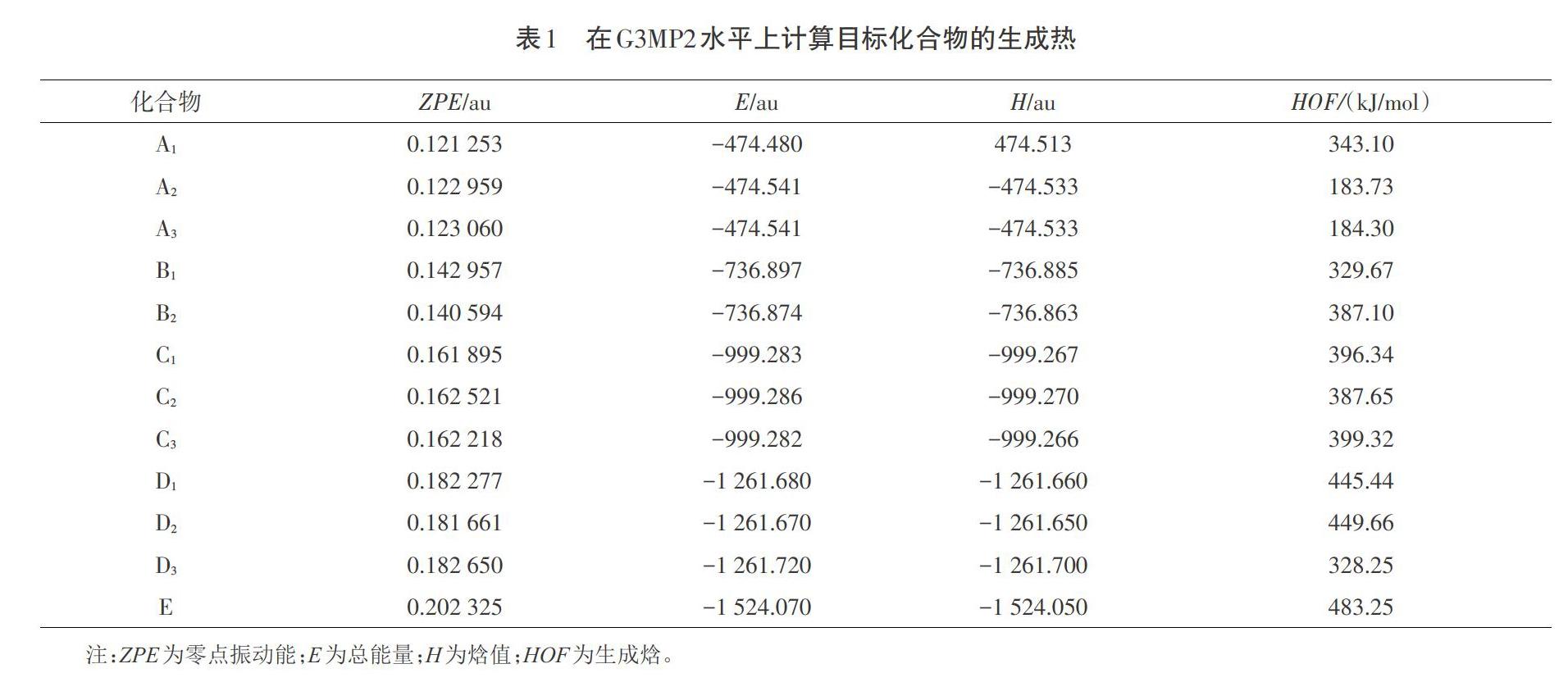
生成热是评价高能密度化合物稳定性和能量含量的一个参数。本文采用等键反应计算的结果列在表1中。表中,A1为1-硝基亚胺吡咯-1H-吡咯;A2为2-硝基亚胺吡咯-1H-吡咯;A3为3-硝基亚胺吡咯-1H-吡咯;B1为1,2-二硝基亚胺吡咯-1H-吡咯;B2为1,3-二硝基亚胺吡咯-1H-吡咯;C1为1,2,3-三硝基亚胺吡咯-1H-吡咯;C2为1,2,4-三硝基亚胺吡咯-1H-吡咯;C3为2,3,4-三硝基亚胺吡咯-1H-吡咯;D1为1,2,3,4-四硝基亚胺吡咯-1H-吡咯;D2为2,3,4,5-四硝基亚胺吡咯-1H-吡咯;D3为1,2,3,5-四硝基亚胺吡咯-1H-吡咯;E为1,2,3,4,5-五硝基亚胺吡咯-1H-吡咯。
所有的生成热都是正值,这是高能密度化合物的典型特征,表明了目标化合物的含能本质。本研究在取代基数和HOFs之间没发现有明显的线性关系。这种情况可能是氢键效应导致的,但是其总体呈现上升的趋势。表1计算结果表明,目标化合物的生成热满足高能量密度化合物的基本要求。
2.2 键离解能和键序
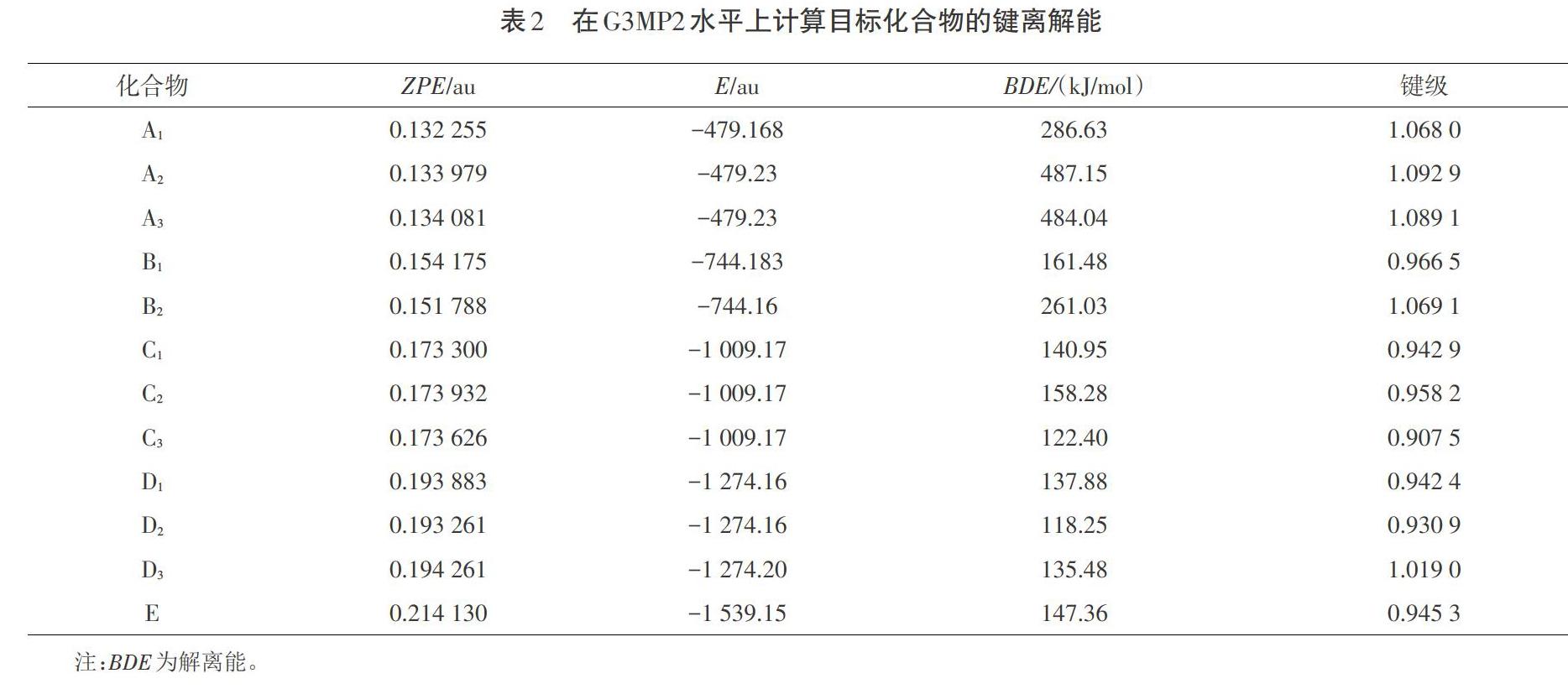
通过计算Wiberg键级,本文最终确定触发键都位于硝氨基基团的N-N键上,其具体结果如表2所示。
从表2可以看出,标题化合物的键离解能很大,最大值是A2的487.15 kJ/mol,最小值是D1的137.88 kJ/mol。与RDX的键离解能(145.62 kJ/mol)相比,标题化合物的键离解能大得足以使化合物在动力学上稳定。键级与键离解之间也没有明显的线性关系。众所周知,键序在试验中是不能被检测到的,因此键离解能是预测分子稳定性的一个较为可靠的参数。
2.3 爆轰特性
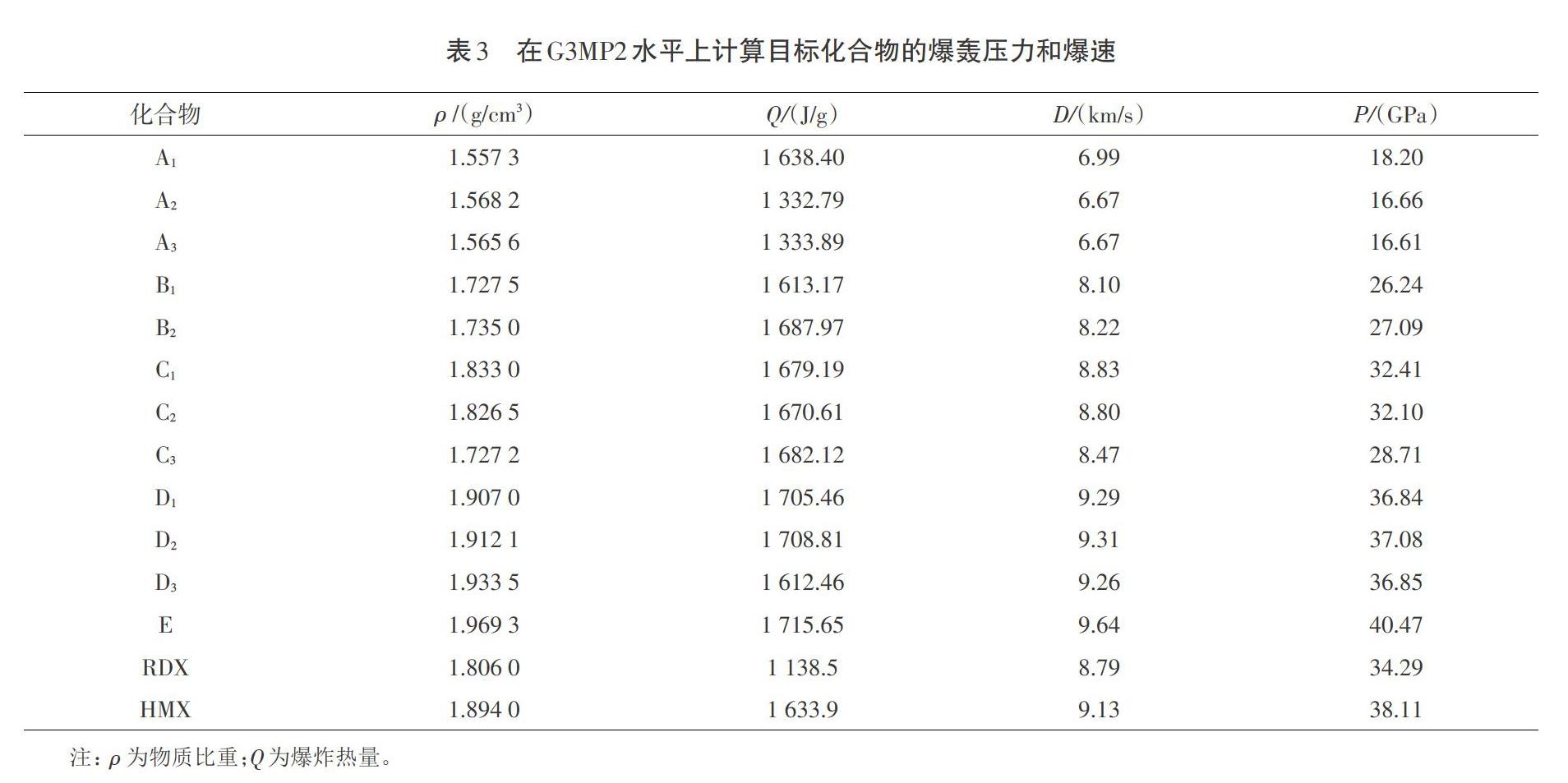
爆轰特性是高能量密度分子的确定性特征之一。最终数据与两个常见的炸药分子RDX和TATB的参数数据也一并列于表3中。
从表3可以明显看出,四取代衍生物和五取代衍生物具有优良的爆炸压力和爆速,前者完全超过了RDX炸药的爆速(8.79 km/s),后者完全超过了RDX炸药的爆压(34.29 GPa)。长期以来,比重大于2.0 g/cm3是下一代高能密度化合物的基本条件,而吡咯衍生化合物的最大比重为1.969 3 g/cm3,爆轰压力为40.47 GPa,爆速为9.64 km/s,优于RDX但是低于HMX。显然,较大的比重并不是提高爆轰特性的决定性因素,其关键是利用硝基亚胺设计低比重高能密度化合物的有效基团。
3 结论
从计算结果可以发现,所有目标化合物的生成热都是正的,满足了高能密度化合物的基本要求。此外,计算出的生成热大于RDX的数值,表明了目标化合物的含能本质。所有衍生物的键离解能都优于RDX和HMX,表明吡咯衍生物具有优秀的动力学稳定性。所有衍生物具有大的爆轰压力和爆速,其中,吡咯的四取代衍生物和五取代衍生物的爆轰压力和爆轰压力均优于RDX和HMX,可以确定为潜在的高能密度化合物。除此之外,计算结果表明,硝基亚胺基团可以改善低比重化合物的爆轰性能。
参考文献:
[1]KUMAR B,Rao M S,KUMAR P,et al.Spectrophotometric investigation of 5-nitroso-6-aminouracil and its methyl derivative in methanol by selective complexation with bivalent metal ions[J].Journal of Molecular Structure,2020(1221):128827.
[2]SHYAMALA B,LAL S,CHOWDHURY A,et al.Cubane decomposition pathways – A comprehensive study[J].Combustion and Flame,2018(197):111-119.
[3]MENG T,WEI J C,QUAN S L, et al.Theoretical design of highly energetic poly-nitro cage compounds[J].RSC Advances,2016(6):47607-47615.
[4]NABATI M,MAHKAM M.Computational Study of Octasilacubane Structural Properties with Density Functional Theory Method[J].Silicon,2016(3):461-465.
