原子替位掺杂对单层Janus WSeTe电子结构的影响*
2021-05-14张德贺周文哲李奥林欧阳方平3
张德贺 周文哲 李奥林 欧阳方平3)†
1) (中南大学物理与电子学院, 长沙 410012)
2) (中南大学粉末冶金研究院, 粉末冶金国家重点实验室, 长沙 410083)
3) (新疆大学物理科学与技术学院, 乌鲁木齐 830046)
基于第一性原理计算系统地研究了氮族、卤族和3d 过渡金属元素(Ti, V, Cr, Mn, Fe, Co)替位掺杂对单层Janus 过渡金属硫族化合物WSeTe 电子结构的影响.通过对能带结构、电荷转移以及磁性的分析, 发现氮(卤)族原子替位掺杂单层WSeTe 会发生本征半导体-p (n)型半导体的转变, Ti, V 原子替位掺杂单层WSeTe会发生半导体-金属的转变.由于电荷转移以及氮族原子掺杂时价带顶的能带杂化现象, 卤族和氮族非金属元素掺杂时价带顶G 点附近的Rashba 自旋劈裂强度在同一主族随着掺杂原子原子序数的增大而增大.3d 过渡金属元素掺杂会产生能谷极化和磁性, 其中Cr, Mn 原子替位掺杂会产生高于100 meV 的能谷极化, 并且Cr,Mn, Fe 元素掺杂在禁带中引入了电子自旋完全极化的杂质能级.研究结果对系统地理解单层WSeTe 掺杂模型的性质具有重要意义, 可以为基于单层WSeTe 的电子器件设计提供理论参考.
1 引 言
石墨稀、六方氮化硼(h-BN)、过渡金属硫族化合物(TMDs)、过渡金属氧化物和磷稀等二维材料由于其卓越的性能和丰富的物理化学性质[1−5], 为其应用和基础研究提供了可能性, 在太阳能电池、晶体管(场效应晶体管)、发光二极管、光电探测器、激光等新型电子和光电子器件中有非常理想的应用前景[6−11].在这些二维材料中, TMDs 因其可调节的带隙以及独特的电子、光学和机械性能而备受关注[12].Janus 过渡金属硫族化合物由于两种硫族原子的电负性以及它们与过渡金属原子之间的键长存在差异使得二维材料产生了面外非对称的电势梯度, 这种面外电势梯度的不对称性引入了Rashba 自旋劈裂[13,14].与其他单层的Janus 过渡金属硫族化合物相比, 研究表明WSeTe 具有更大的自旋劈裂[15], Janus 过渡金属硫族化合物独特的性质使其在压电效应和自旋电子器件等诸多领域具有很好的应用前景.
通过化学气相沉积法, 用Se 原子完全取代了MoS2的最上层所有S 原子, 成功地合成了极性MoSSe[16].除此之外, 通过自下向上和自上向下的方法, 使用机械剥离、液相剥离、物理气相沉积、溶液化学过程和化学气相输运[17−20]等方法都可以成功制备Janus 二维过渡金属硫族化合物.已有的工作对单层Janus 过渡金属硫族化合物在光催化剂、电子结构、磁性、声子输运以及析氢反应的催化[21−25]等方面都进行了广泛的研究.原子替位掺杂是一种改变低维体系电子结构十分简单且有效的方式.例如Cd 掺杂使Bi2Se3薄膜由n 型半导体到p 型半导体的转变[26].在石墨烯中掺杂B 和N 原子在狄拉克点附近打开带隙[27].3d 过渡金属元素掺杂在非磁的二维材料中引入磁性[28,29]等.
本文通过第一性原理计算系统地分析了VA(N, P, As, Sb)和VIIA (F, Cl, Br, I)非金属元素替换一个Se 原子以及3d 过渡金属(Ti, V, Cr, Mn,Fe, Co)元素替换一个W 原子掺杂单层WSeTe超胞对电子结构的影响.VA (VIIA)非金属元素掺杂会引入空穴(电子); 3d 过渡金属元素掺杂会破坏时间反演对称性从而引入磁性和能谷极化现象[30].
2 计算模型与方法
本文使用基于密度泛函理论(DFT)的VASP软件[31,32]包进行计算, 采用投影缀加平面波[33]和广义梯度近似下的Perdew-Burke-Ernzerhof(PBE)[34]泛函的方法同时设置截断能为400 eV.为了消除周期性边界条件下其他原子对单层WSeTe 的影响, 在Z轴方向上设置了一个20 Å的真空层.能量的收敛标准设为10–5eV, 力的收敛标准设为0.02 eV/Å.利用Monkhorst-Pack[35]方法对第一布里渊区进行采样, 由于超胞较大布里渊区K点网格设置为4 × 4 × 1.在过渡金属元素替位W 原子掺杂的计算中, 加入了自旋轨道耦合(SOC)并采用DFT +U的方法(Hubbard 参数Ueff= 3 eV)[36].
3 计算结果与讨论
首先建立了一个单层WSeTe 六角晶系的4 ×4 超胞含有48 个原子.在VA (VIIA)元素替位掺杂单层WSeTe 时将其中的一个Se 原子替换为氮族原子(卤族原子); 在3d 过渡金属(TM)元素替位掺杂单层WSeTe 时将其中的一个W 原子替换为过渡金属原子, 掺杂浓度为2.08%, 对x,y方向的晶格常数进行了优化, 所有结构都经过了充分弛豫.如图1(c)所示, 一个没有掺杂的4 × 4 WSeTe超胞导带底和价带顶都处于K点, 是一个带隙为0.98 eV 的直接带隙半导体, 与文献上计算得到的原胞能带结构符合较好[15].
3.1 VIIA (VA)元素替位掺杂单层WSeTe
首先对VIIA (VA)元素替位Se 原子掺杂单层WSeTe 的结构进行了优化, 表1 的结果表明Cl, Br, I, N, P, As, Sb 掺杂后具有稳定的几何结构以及低的形成能Ef.
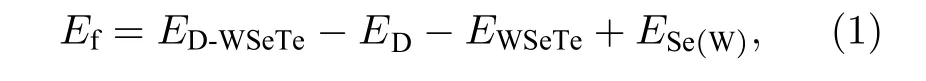
其中ED-WSeTe是原子替位掺杂WSeTe 后的能量,ED是一个孤立掺杂原子的能量,EWSeTe是WSeTe超胞的能量,ESe(W)是一个孤立的Se (W)原子能量.F 原子半径较小且电负性较强使得F 原子更多是以间隙掺杂或吸附的形式存在于WSeTe 中, 因此F 原子替位掺杂单层WSeTe 无法优化得到一个稳定的结构.随着原子序数的增大, VIIA (VA)掺杂原子与W 原子之间的键长dW−D会逐渐增加.
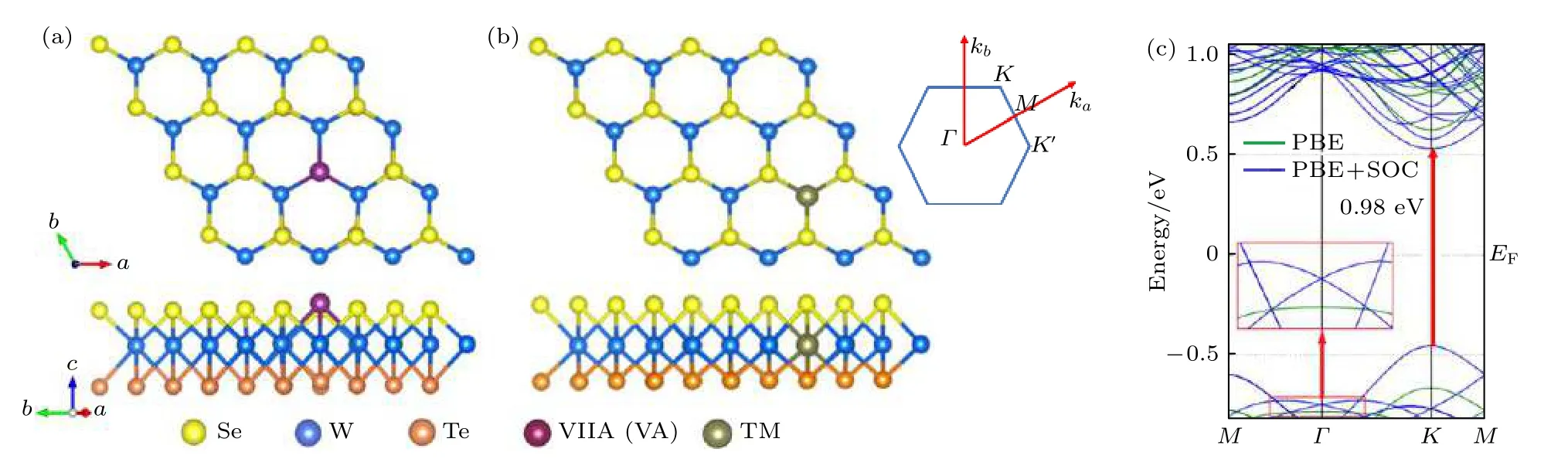
图1 (a) VA (VIIA)和(b) TM 元素替位掺杂单层WSeTe 的俯视图(上)和侧视图(下)及其布里渊区和高对称点示意图;(c) 4 × 4 的单层WSeTe 超胞的能带结构Fig.1.(a), (b) The top view (top) and the side view (bottom) of the substitutionally doped monolayer WSeTe of VA (VIIA) and TM elements, as well as the schematic diagram of Brillouin zone and high symmetry points; (c) the energy band structure of 4 × 4 monolayer WSeTe supercell.
VIIA 元素替位掺杂单层WSeTe 时由于卤族元素最外层比Se 原子多了一个电子, 因此卤族元素替位掺杂WSeTe 相当于引入了电子掺杂并且能带中的费米能级上移到了原本导带底的位置使得材料变为n 型半导体.图2(a)中使用PBE + SOC方法得到的掺杂后的能带结构仍然保留了较宽的禁带, 只有导带底K点附近较窄的区域穿过费米能级.通过对比Cl, Br, I 掺杂后的能带结构图可以发现, 随着原子序数的增大, 加入SOC 得到的能带中最高占据态(最低未占据态)会略微上(下)移, 减小禁带宽度.从单个原子的投影态密度(PDOS)图2(d)可以看出, 费米能级附近能带主要来源于材料本身W 原子的贡献.由于掺杂浓度较低且掺杂原子对费米能级附近能带贡献较小, 不同的卤族元素掺杂时能带结构并没有明显的变化.为了了解VIIA 元素掺杂过程中电荷的转移情况,计算了它们的差分电荷密度图, 从图2(e)可以发现, 电荷转移主要发生在卤族原子和与其最近邻的3 个W 原子中, 并且与掺杂原子最近邻的3 个W 原子转移的电荷是完全相同的, 而Se 原子和Te 原子以及其他位置的W 原子在卤族元素替位掺杂前后并没有发生比较明显的电荷转移.
VA 元素掺杂单层WSeTe 时由于氮族元素最外层比Se 原子少了一个电子, VA 元素替位掺杂单层WSeTe 相当于引入了空穴掺杂并且能带中的费米能级向下移动到了原本价带顶的位置使得材料变为p 型半导体.通过对比图3 的能带结构可以看出, N, P 原子替位掺杂单层WSeTe 时费米能级附近的能带更加局域, 在WSeTe 的禁带中引入了杂质能级.如图3(b)和图3(d)所示, 通过计算单个原子的PDOS 发现, 在费米能级附近掺杂原子对能带的贡献随着原子序数的增大而减小, 因此N, P 掺杂单层WSeTe 得到的能带在费米能级附近杂化现象更强, 而As, Sb 原子掺杂单层WSeTe由于杂化现象较弱得到的能带与本征的单层WSeTe 能带结构在价带顶差异较小, 尽管如此我们仍然可以看到对于加入SOC 的能带, As, Sb 原子掺杂对材料的电子结构尤其是价带顶的Rashba自旋劈裂仍然产生了很大的影响.对于单个原子而言, Sb 掺杂单层WSeTe 时费米能级附近的能带主要来源于W 原子和掺杂原子的贡献; 而对于原子序数较小的N 掺杂单层WSeTe, 在费米能级附近Se 原子和Te 原子对能带也具有相当大的贡献.
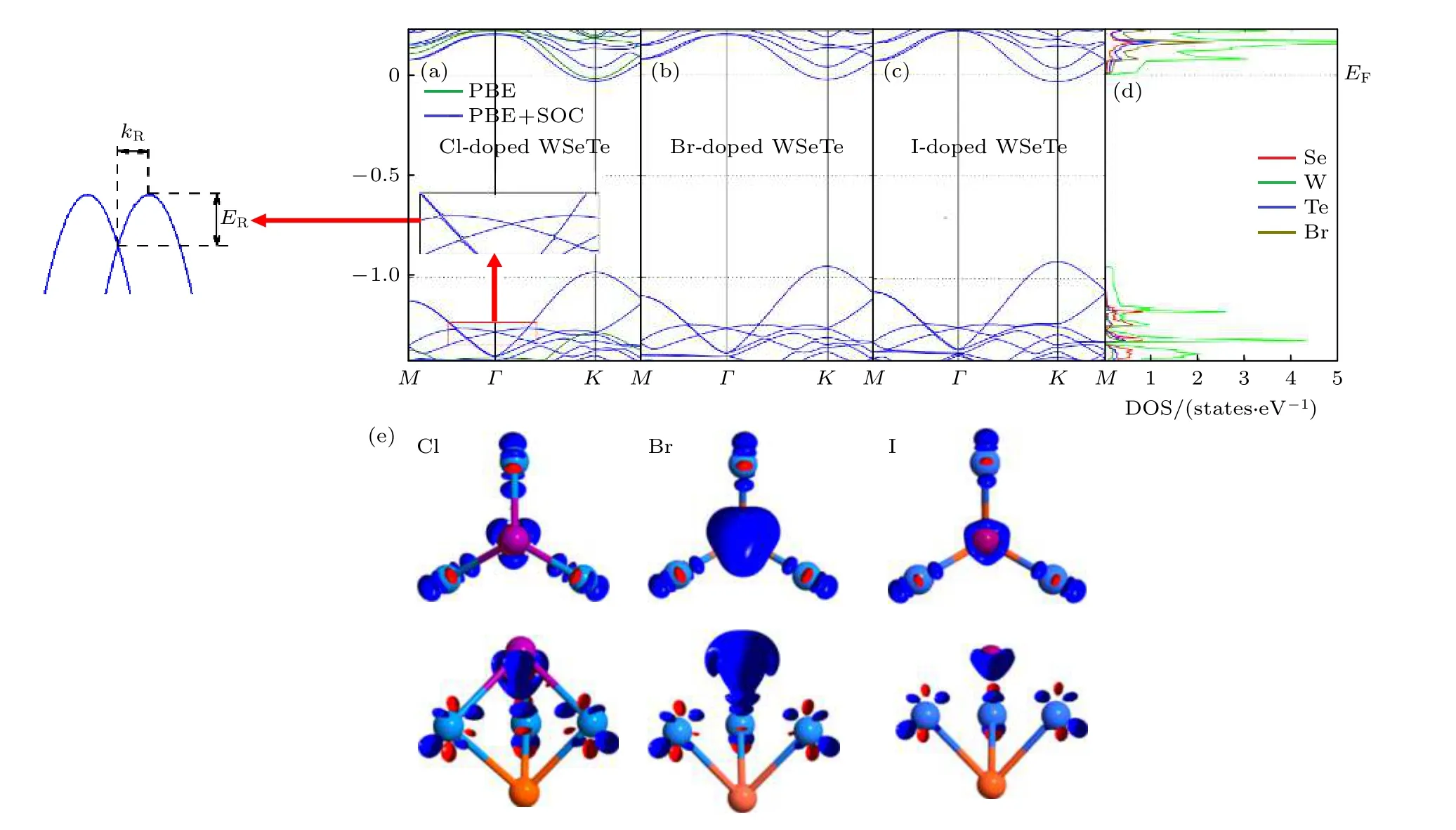
图2 (a), (b), (c) Cl, Br, I 原子掺杂单层WSeTe 能带结构图; (d) Br 原子掺杂单层WSeTe 中单个原子的投影态密度图; (e) VIIA原子掺杂单层WSeTe 的差分电荷密度的俯视图(上)和侧视图(下), 红色代表等值面0.01 a.u., 蓝色代表等值面–0.01 a.u.Fig.2.(a), (b), (c) The WSeTe band structure of Cl, Br and I atom doped, respectively; (d) the project density of state for each single atom in monolayer WSeTe doped by Br atom; (e) the top view (top) and side view (bottom) of the differential charge density of a monolayer WSeTe doped with VIIA atoms, red represents the isosurface 0.01 a.u.and blue represents the isosurface–0.01 a.u..
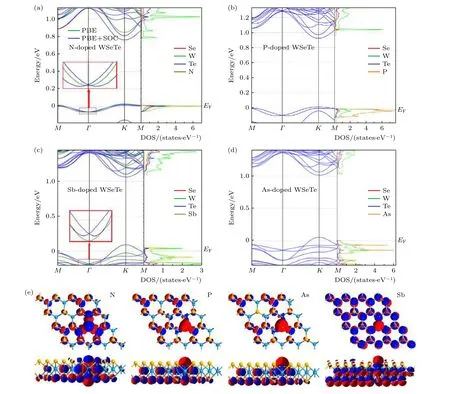
图3 (a), (b), (c), (d) N, P, Sb, As 掺杂单层WSeTe 的能带结构图和单个原子的PDOS 图; (e) VA 原子掺杂单层WSeTe 的差分电荷密度的俯视图(上)和侧视图(下), 红色代表等值面0.01 a.u., 蓝色代表等值面–0.01 a.u.Fig.3.(a), (b), (c), (d) The band structure diagram of N, P, Sb and As doped single-layer WSeTe and the PDOS diagram of a single atom, respectively; (e) the top view (top) and side view (bottom) of the differential charge density of a monolayer WSeTe doped with VA atoms, red represents the isosurface 0.01 a.u and blue represents the isosurface –0.01 a.u..
为了更加直观地了解电荷的转移情况, 计算了VA 元素掺杂单层WSeTe 的差分电荷密度.如图3(e)所示, VA 元素掺杂二维WSeTe 时电荷转移不仅仅只发生在VA 原子和与其最邻近的3 个W 原子之间, 其他的W 原子和Te 原子也有一部分会发生明显的电荷转移, 这是由于卤族原子电负性较强, 与最邻近的过渡原子吸引电子能力差距较大且最外层电子达到饱和状态需要的电子数少, 最近邻的W 原子能够提供卤族原子所需的电子数因此电荷转移较为集中; 而氮族元素的电负性较弱,与过渡原子吸引电子能力的差距较小且最外层电子达到饱和状态需要的电子数多, 最近邻的W 原子不能提供氮族原子所需的电子数, 因此各原子间的电荷转移较为分散.同时可以观察到远离掺杂原子区域的电荷转移并不是发生在不同原子之间的,而是从原子的一侧转移到相反的另一侧, 电荷分布产生了一定的极化, 但单个原子本身总的电荷数量并没有明显的变化.
研究了VIIA (VA)非金属元素替位掺杂对G点附近Rashba 自旋劈裂强度的影响, 通常在Rashba 自旋劈裂效应中决定自旋劈裂强度的Rashba 常数被定义为
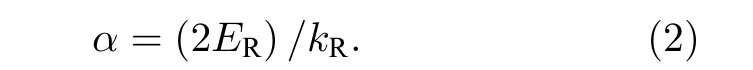
在图2(a)的示意图中ER为自旋劈裂能量,kR为偏移动量.由于单层WSeTe 在导带底Rashba自旋劈裂较小, 主要考虑其价带顶的Rashba 自旋劈裂.如图1(c)所示, 通过对比加入SOC 和不加入SOC 的能带结构, 可以看到WSeTe 价带顶的能带加入SOC 之后在G点劈裂成了两条能带.由于Rashba 常数对布里渊区的方向选择并不敏感[37], 使用G—K方向来计算α, 计算的结果显示在表1 中.
从图1(c)和图2 可以看出, 当VIIA 元素替位掺杂WSeTe 时,G点附近价带顶处的能带与本征的WSeTe 相同都是由两条开口向下的抛物线组成的; 而从图3(a)和图3(c)可看出, VA 元素替位掺杂WSeTe 时,G点附近价带顶处的能带是由两条开口向上的抛物线组成的.这是由于VA 元素掺杂后价带顶的能带具有较强的杂化, 价带顶G点附近的自旋劈裂是杂质原子与过渡原子之间的SOC所引起的, 这种SOC 强度随氮族元素原子序数的增加而增强.VIIA 元素替位掺杂WSeTe 时掺杂的卤族元素对价带顶几乎没有贡献, 因此VA 元素替位掺杂WSeTe 时Rashba 常数α随原子序数的变化幅度远大于VIIA 元素替位掺杂.从表1 可以发现, VIIA 元素替位掺杂WSeTe 时Rashba 常数α随着掺杂原子的原子序数的增大而增大, 这是由于同一主族元素随着原子序数的增大掺杂原子得到的电子数目会相应减少, W 原子保留的电子数目增加并且价带顶G点附近的Rashba 自旋劈裂主要是由W 原子贡献, 因此会增大Rashba 常数α,当然这也是VA 元素替位掺杂WSeTe 时Rashba常数α随着掺杂原子的原子序数的增大而增大的原因.当考虑电负性时, 会发现原子序数小具有较强电负性的原子掺杂在电负性强的Se 原子一侧时, 理论上会增强两侧的电负性的差异从而增强Rashba SOC 强度, 但得到的结果却恰恰相反.这说明原子序数增大以及电荷转移对Rashba SOC强度的影响要大于电负性的变化产生的影响.从之前报道的Mo(W)SeTe 中的Rashba SOC 强度远大于Mo(W)STe[38]中也能得到相似的结论.
3.2 过渡金属元素替位掺杂单层WSeTe
用V, Cr, Mn 等本身具有磁性的元素完全取代Janus 过渡金属硫族化合物的Mo(W)原子会产生较大的自旋极化和较高的居里温度[38], 但这些材料都是金属或半金属性质不利于半导体器件的应用.因此我们想通过部分掺杂过渡金属元素来保留材料的半导体性质并获得较好的磁性和自旋极化性质.在超胞中替位掺杂过渡金属元素结构上仍然保持原本的对称性, 优化的结构稳定具有低的形成能.
计算了Ti, V, Cr, Mn, Fe, Co 过渡金属元素替位W 原子掺杂单层WSeTe 的能带结构, 从图4可以看出, 价带顶自旋方向相反的电子产生了能谷极化现象, 从表2 可以发现, Cr, Mn, V 元素掺杂会产生较大的能谷极化.以Ti 掺杂为例, 过渡金属掺杂在图1 中的高对称点K和K'谷处对自旋相同的电子产生相同的自旋劈裂能量, 而SOC 在K和K'谷处对自旋相同的电子产生相反的自旋劈裂能量, 使得SOC增加了K谷的自旋劈裂能量,减小了K'谷的自旋劈裂能量, 所以K谷的自旋劈裂大于K'谷[39].其中掺杂Ti, V 元素会导致体系由半导体向金属的转变, 掺杂Co, Cr, Mn, Fe 元素不会导致材料的半导体性质发生变化, 但Co,Mn, Fe 元素掺杂后的能带具有极小的带隙(小于20 meV), 不过可以通过调控费米能级或带隙的方法使材料变为半金属或宽带隙半导体, 从而获得更好的应用前景.而Cr 元素掺杂WSeTe 由于其很好地保留了WSeTe原本的半导体性质并且具有较大的能谷极化, 在自旋电子器件领域具有很好的应用前景, 遗憾的是Cr 元素掺杂WSeTe 能带的价带顶仅仅在布里渊区K点附近很小的区域有自旋极化向下的电子.Ti, V 元素掺杂的体系虽然是金属性质但在最高轨道占据态的K和K'点周围大范围内全部由自旋向下(上)的电子所占据, 拥有大量可调控的自旋电子.除了能谷极化现象外, 由于过渡元素的掺杂破坏了原本的周期性势场, Cr,Mn, Fe 元素掺杂单层WSeTe 会在原本的禁带中产生自旋电子完全极化向下的杂质能级, 杂质能级之下的能带是由自旋极化向上的电子构成, 可以通过外加电场等方式调控费米能级位置来达到自旋翻转的效果, 这对自旋电子的调控具有重要意义.
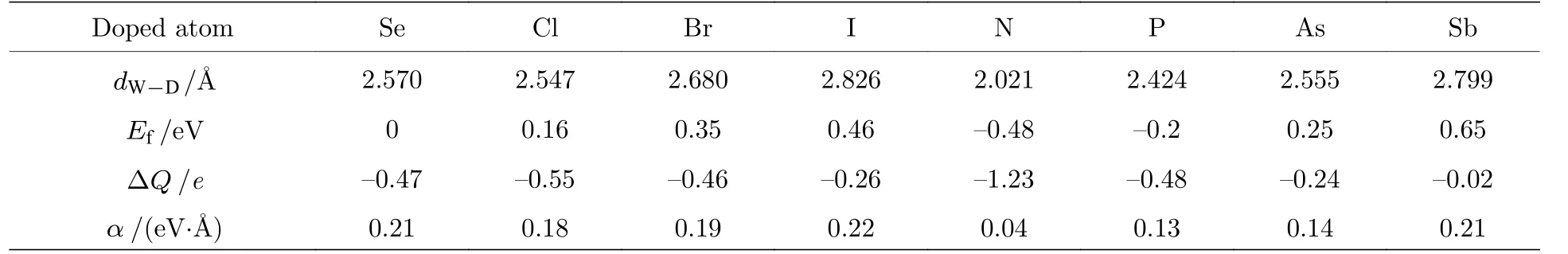
表1 未掺杂体系的Se 原子或VA (VIIA)掺杂原子和W 原子之间的键长 d W−D 、体系形成能 E f 、掺杂原子得到的电荷数 Δ Q 以及Rashba 常数αTable 1.Bond length d W−D between VA (VIIA) doped atoms or Se atoms in an undoped system and W atoms, binding energy E f , the number of charges obtained by doping atoms Δ Q and Rashba constant α.
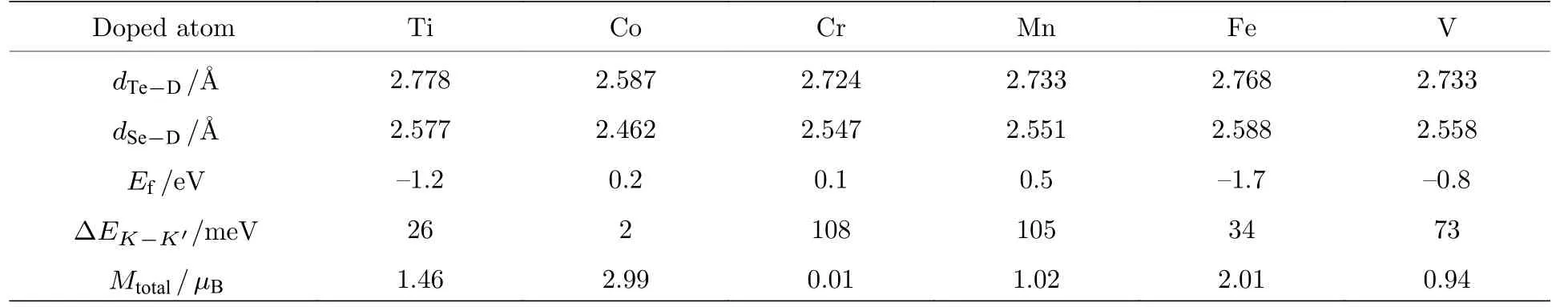
表2 掺杂原子与Se (Te) 原子之间的键长 d Se−D ( d Te−D )、结合能 Ef 、能谷极化的大小 Δ EK−K′ 及体系总的磁矩MtotalTable 2.Bond length d Se−D(dTe−D) between doped and Se (Te) atoms, binding energy E f , energy valley polarization ΔEK−K′ and total magnetic moment M total.
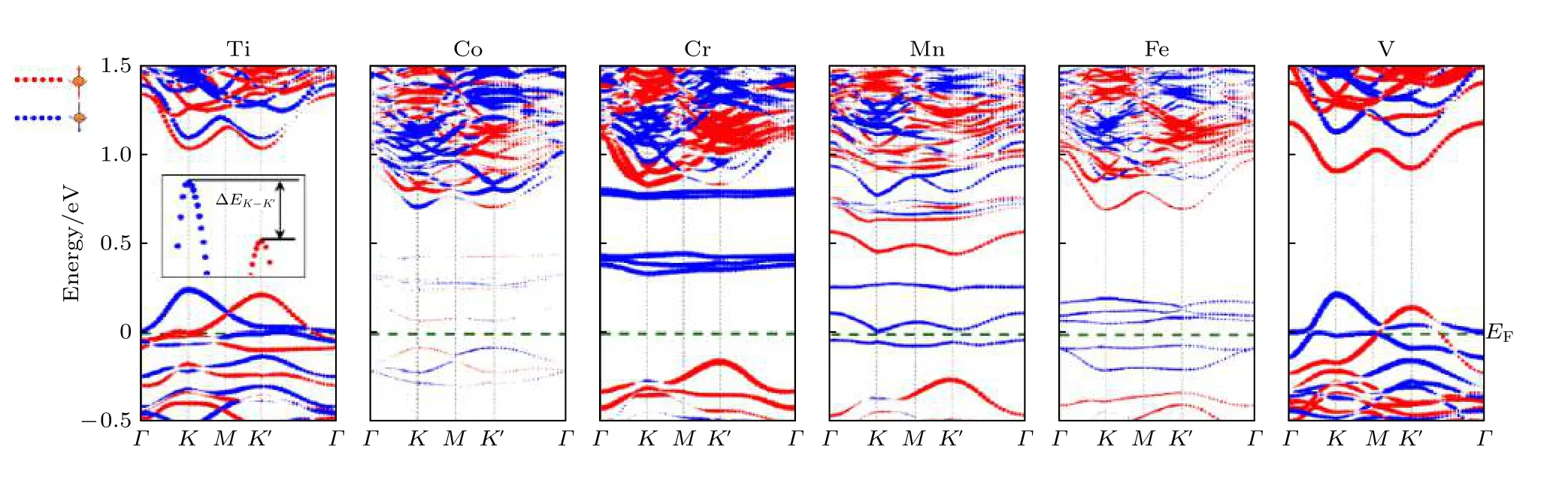
图4 3d 过渡金属元素掺杂单层WSeTe 的能带图, 自旋在z 轴方向的期望值 〈 Sz〉 的正负分别用红色和蓝色来表示, 〈 Sz〉 绝对值的大小用点的大小来表示; 所有的能带经过平移使费米能级对齐, 同时费米能级固定在0 eV 处Fig.4.Band diagram of 3d transition metal elements doped monolayer WSeTe.The positive and negative values of the expected value 〈 Sz〉 of the spin in the z-axis direction are represented by red and blue respectively, and the magnitude of the absolute value of 〈 Sz〉 is represented by the magnitude of the points.The spin projection along z-direction is depicted by the magnitude of the point.All the bands are shifted to align the Fermi level which is fixed at 0 eV.
由于Ti, V 元素金属性较强使得Ti, V 元素掺杂的体系表现为明显的金属性, 而Co, Cr, Mn,Fe 元素掺杂体系仍为半导体性质.Co 元素掺杂时产生的磁矩主要沿面内方向, 因此体系的能谷劈裂程度很小.Cr 元素掺杂时掺杂的Cr 原子具有3µB的磁矩, 但被其附近的原子所产生的自旋方向相反的磁矩抵消, 体系总磁矩为0, 因此Cr 掺杂时体系仍具有能谷劈裂现象.Fe 元素掺杂时杂质能级穿过了价带顶, 破坏了能谷极化现象.能谷劈裂是体系本身的自旋劈裂与掺杂原子引入的磁性共同作用的结果, 不同体系中能量较高(低)的能谷由不同自旋占据, 仅仅是由于体系磁矩沿面外的方向不同, 通过外加z方向的磁场可以改变体系磁矩沿面外的方向, 从而翻转高(低)能谷的电子自旋.
由于过渡金属元素替位掺杂单层WSeTe 破坏了材料原本的时间反演对称性, 在表2 中Ti, V,Mn, Fe, Co 原子替位掺杂WSeTe 都产生了一定的磁矩.由于Ti 和V 原子掺杂WSeTe 是金属性质, 费米能级附近具有巡游的3d 电子, 这部分电子对磁矩也有贡献, 使得产生的总磁矩不是整数,分别为1.46 和0.94µB.Cr 与W 原子由于电子结构的相似, 替位掺杂不产生磁矩, 在对磁矩组成部分的分析发现, 磁矩主要来源于掺杂原子和材料本身W 原子未填满的d 轨道电子.
4 结 论
通过以DFT 为基础的VASP 软件包计算了VA (VIIA)非金属元素替位Se 原子以及3d 过渡金属元素替位W 原子掺杂单层WSeTe 超胞对电子结构的影响.VA (VIIA)非金属元素替位Se 元素掺杂单层WSeTe 由于引入了空穴(电子)掺杂,使得费米能级下移(上移), 材料变为p (n)型半导体.Ti, V 元素替位掺杂会发生半导体-金属的转变, 替位掺杂Cr, Co, Mn, Fe 元素不会导致材料的半导体性质发生变化, 但Co, Mn, Fe 元素掺杂后的带隙小于20 meV.VIIA (VA)非金属元素和3d 过渡金属元素掺杂单层WSeTe 不会对材料的几何结构产生巨大的影响.由于电荷转移以及VA 掺杂原子在价带顶的能带杂化现象, VIIA(VA)非金属元素掺杂时对于同一主族元素, 价带顶的Rashba 自旋劈裂强度随着掺杂原子的原子序数的增大而增大.3d 过渡金属元素替位掺杂单层WSeTe 具有明显的自旋极化现象, 在费米能级附近产生了能谷极化并引入磁性, Cr, Mn, Fe 元素掺杂单层WSeTe 在禁带中产生了自旋电子完全极化的杂质能带, 在自旋电子器件方面可能有潜在的应用价值.研究结果对系统地理解单层WSeTe掺杂模型的性质有重要意义, 可以为基于单层WSeTe 的电子器件设计提供理论参考.
