Fe3O4@MnOx介孔球形材料的制备及其吸附U(VI)的研究
2021-05-12张婷婷郑驯梅金家宏胡保卫
张婷婷 胡 忻 郑驯梅 金家宏 胡保卫
(1.绍兴文理学院 土木工程学院,浙江 绍兴 312000;2.绍兴文理学院 生命科学学院,浙江 绍兴 312000)
铀(U)是核能运行中的主要燃料,但铀矿开采冶炼的过程中会产生大量的矿渣和尾水,对生态环境造成放射性污染[1-2].U在水体中主要以易迁移的六价U(VI)存在.目前处理U(VI)污染场地常用的方法有物理法、化学法和生物法[3-6],其中物理法中的吸附法因操作简单、效率高而被广泛采用[7].吸附法的关键是吸附材料的选择,材料不仅要求吸附容量大、速度快,还需要具有易回收和循环利用率高等特点.
近年来,介孔材料因比表面积大,具有介孔孔道等优点在U(VI)的去除中受到越来越多的关注,但是由于其密度小、分散度高,在水体中易迁移流动,吸附时容易对水体环境造成二次污染.在介孔材料上负载Fe3O4制备成磁性介孔材料,既保留了介孔的优良特性,也可以实现材料的快速分离回收,大大增加了其在吸附领域的实用性[8-9].MnOx是一种结晶性较差的无定型结构的氧化物,比表面积大,对U(VI)具有高效地吸附能力,在磁性介孔材料上负载MnOx,既可以通过Mn-O (Mn-OH)与U(VI)的结合提高吸附剂的吸附能力,也可以解决磁性材料易被氧化的问题[10,11].
因此本文拟通过模板法和溶剂热法合成Fe3O4@MnOx磁性介孔材料,并进行一系列的表征和吸附试验,研究其对水体中U(VI)的吸附能力,为放射性污染问题建立经济、环保、高效的修复方式以及在新材料和新技术方面提供必要的理论依据.
1 材料与方法
1.1 实验材料与实验设备
聚乙烯氧化物-聚丙烯氧化物-聚乙烯氧化物(P123),二茂铁(Fe(C5H5)2),正硅酸四乙酯(TEOS)购于阿拉丁.乙醇,过氧化氢(H2O230% w/w),高锰酸钾(KMnO4)购于上海凌峰化学试剂有限公司.硝酸铀酰(UO2(NO3)2·6H2O)购自湖北楚盛威化工有限公司.实验用水均为超纯水,所有试剂均为分析纯.采用日本电子公司的Tecnai G220型透射电子显微镜(TEM)和JSM-6360LV型扫描电子显微镜(SEM)观察材料的表面形貌.通过英国牛津X-act的能谱(EDS)进行元素分析.用荷兰帕纳科Empyrean X-射线衍射仪(XRD)测定晶体结构和物相.通过美国的Micromeritics公司的比表面积和孔隙度分析仪(SAP)测定样品比表面积以及孔径范围.美国Thermo Scientific公司ESCALAB-250Xi型X-射线能谱(XPS)定性分析材料中铁锰的价态.英国马尔文仪器有限公司的零点电位分析仪测定样品的Zeta电位.
1.2 Fe3O4@MnOx介孔球形材料的制备
该材料采用模板法和溶剂热法合成,制备过程如下:取4 g P123溶于90 mL的2 M HCl中,并加入21 mL去离子水,常温搅拌5 h;加热至40 ℃后加入6.4 g TEOS缓慢搅拌持续24 h;所得悬浮液移至水热反应釜中,100 ℃下晶化24 h.常温下冷却后,混合物用丙酮多次洗涤并抽滤,在80 ℃的烘箱中干燥2 h,然后将所得的白色固体粉末置于600 ℃的马弗炉中煅烧6 h,得到介孔硅基模板SBA-15.称取200 mg SBA-15与200 mg二茂铁分散于65 mL丙酮中,猛烈搅拌45 min,搅拌过程中缓慢滴加2 mL H2O2,随后移至水热反应釜中,200 ℃反应20 h.冷却后离心,得到的固体用丙酮和乙醇反复洗涤后在60 ℃下干燥12 h得到Fe3O4/C材料.将200 mg Fe3O4/C材料溶于70 mL的KMnO4(0.005 M)溶液.在160 ℃反应12 h.用永久磁铁分离得到固体,所得固体用3 M的NaOH溶液洗涤,再用去离子水反复清洗,于60 ℃过夜真空干燥,得到Fe3O4@MnOx介孔球形材料.
1.3 吸附实验
配制浓度为50 mg/L的U(VI)溶液,用体积可以忽略不计的HCl和NaOH调节pH.取100 mL溶液置于250 mL锥形瓶中,加入0.1 g吸附剂后置于恒温摇床中以150 rpm的速度震荡,定时取样,分析pH对吸附的影响.在pH=5的100 mL溶液中加入0.1 g Fe3O4@MnOx进行吸附动力学实验.控制温度变化,调节pH至5,在100 mL的U(VI)溶液中加入0.1 g Fe3O4@MnOx,研究温度对U(VI)吸附的影响及其吸附等温模型拟合.反应后取1 mL样品,过0.45 μm水系滤膜,所得滤液用偶氮胂Ⅲ显色法(波长为652 nm)测定滤液中U(VI)的含量.吸附容量以及吸附率计算公式如下:
(1)
(2)
其中C0(mg/L)为U(VI)溶液的初始浓度,Ce(mg/L)为U(VI)溶液吸附平衡时的浓度;m(g)和V(L)分别为吸附剂的投加量和U(VI)溶液的体积;Sorption(%)为吸附率;qe(mg/g)为平衡时的吸附容量.
2 结果与讨论
2.1 pH对吸附的影响
由于吸附剂的表面电荷和U(VI)在水相中的物种存在形式对U(VI)的吸附有很大的影响,因此有必要进行pH对U(VI)的吸附影响的研究.pH对U(VI)的吸附效果影响以及Fe3O4@MnOx的Zeta电位如图1所示.

(a) pH对U(VI)的吸附影响;

图1 (b) Fe3O4@MnOx的Zeta电位
2.2 吸附动力学分析
固定U(VI)的初始浓度、投加量以及体积,探究接触时间对吸附效果的影响.接触时间对U(VI)吸附效果的影响如图2所示,随着接触时间的增大,吸附容量也快速增加,当接触时间接近30 min时,吸附几乎接近吸附平衡.在实际应用中,吸附速率的高低影响着吸附材料性能的优劣,为了研究Fe3O4@MnOx的吸附动力学,对图2中的吸附数据进行伪一级动力学模型(3)和伪二级动力学模型(4)的拟合[16-17],其相关拟合参数列于表1.

图2 Fe3O4@MnOx吸附U(VI)的吸附动力学模型拟合
(3)
(4)
其中,qe(mg/g)和qt(mg/g)分别为平衡状态时和时间为t时每单位质量吸附剂的吸附量.k1(min-1)和k2(g/mg·min) 分别为伪一阶、伪二阶方程的吸附速率常数.从拟合结果可以看到动力学模型均比较贴近实验值,从拟合的相关系数R2来看,虽然两者数值相差不大,但是伪二级比伪一级能更好地用来描述U(VI)在Fe3O4@MnOx上的吸附过程,也表明U(VI)在Fe3O4@MnOx上的吸附过程不仅是有化学吸附,并且与物理吸附具有协同作用[18-19].从图2中也可以看出U(VI)在Fe3O4@MnOx上的初始吸附是快速吸附,吸附10 min后去除率达到63.7%,初始吸附速度很快,可能是因为Fe3O4@MnOx上有很多可利用的吸附位点,当反应达到30 min时,吸附位点逐渐减少,吸附速率减慢,吸附达到平衡,再继续延长反应时间,吸附效果增加不明显,吸附趋于饱和.

表1 伪一级和伪二级吸附动力学模型的拟合参数和相关系数
2.3 吸附等温模型分析
不同温度下的不同吸附等温模型拟合如图3所示.分别采用Langmuir (5)和Freundlich (6)吸附等温模型[20-21]对Fe3O4@MnOx吸附U(VI)的吸附数据进行拟合,拟合参数如表2所示.
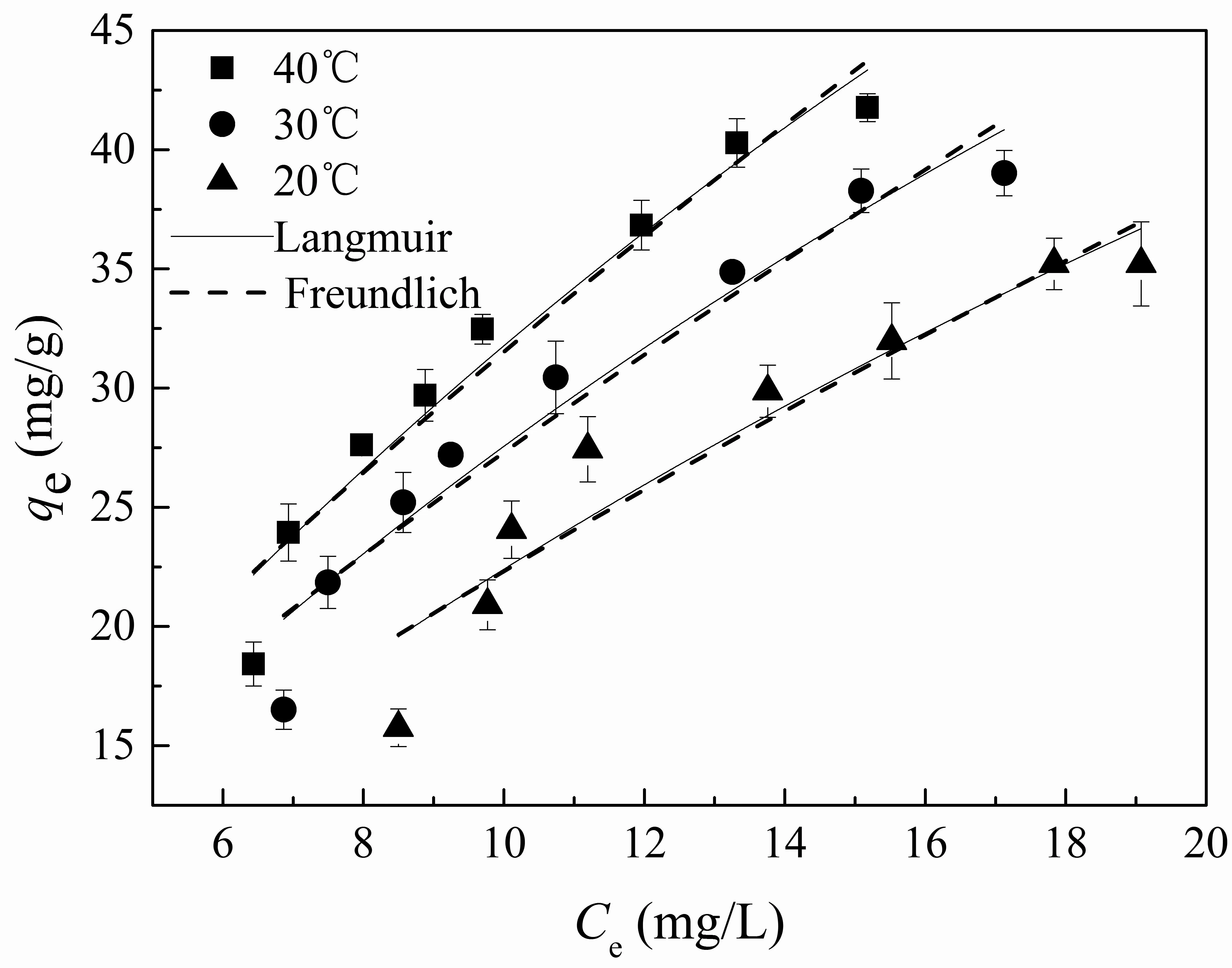
图3 在不同温度下Fe3O4@MnOx吸附U(VI)的吸附等温模型拟合
(5)
(6)
其中,qe(mg/g)为平衡状态时每单位质量吸附剂的吸附量,b(L/mg)为Langmuir的吸附系数,kF(mg1-n·Ln/g)和n是Freundlich的经验常数.Langmuir模型是单层吸附理论模型,Freundlich模型是非均质表面的多层吸附,是一个经验吸附平衡方程.
实验结果表明,吸附容量随反应温度的升高而升高,吸附能力增大,表明Fe3O4@MnOx对U(VI)的吸附高温有利,低温受阻.在不同温度下(20 ℃、30 ℃、40 ℃)的Freundlich吸附等温模型拟合中,1/n<1,这表明U(VI)在实验范围内吸附反应较容易发生.吸附平衡常数kF可以反映吸附容量的大小,kF随温度升高而增大,Fe3O4@MnOx对U(VI)的吸附容量也是随温度的升高而升高.由不同温度下的吸附等温模型拟合出的相关系数R2可知,Langmuir模型更适合用于描述Fe3O4@MnOx对U(VI)的吸附过程.
2.4 Fe3O4@MnOx材料表征
2.4.1 SEM、TEM和SEM-EDS表征
通过扫描电镜(SEM)、透射电镜(TEM)以及扫描电镜-能谱(SEM-EDS)观察Fe3O4@MnOx的表面形貌以及表面元素分布. SEM表征如图4 (a)所示,可以清楚地看到在扫描电镜下Fe3O4@MnOx是由许多表面粗糙的具有介孔结构的球形颗粒组成,大小、尺寸均匀. TEM表征如图4 (b)所示,与嵌入图中未负载MnOx的Fe3O4/C的TEM图相比,可以清楚地看到负载MnOx后的磁性纳米材料具有明显的核壳式结构,表面包裹的一层类似棉絮状的锰氧化物,且吸附材料表现出良好的多分散性和较规则的形状[22],更好地佐证了SEM的结果.从图4 (d)吸附后的SEM图中可以清楚地看到原本松散的球形颗粒吸附后结块,空隙被U(VI)填满,并且从4 (e)的嵌入图中可以看到在外加磁场的作用下Fe3O4@MnOx可以很容易地被聚集在一起,这也说明了Fe3O4@MnOx具有良好的磁性,可以实现回收利用的目的.EDS表征如图4 (c)和4 (f),图4 (c)为未负载MnOx的EDS,嵌入图为负载MnOx后的Fe3O4@MnOx的EDS图,由图可知,负载MnOx后,材料的元素组成主要是Fe、Mn、Si、O.图4 (f)中可以看到,Fe3O4@MnOx吸附剂表面Fe、Mn元素分布均匀,两者含量相差不多,大量的U(VI)已经成功的吸附在了Fe3O4@MnOx上.

表2 Langmuir和Freundlich等温吸附模型的拟合参数和相关系数

(a)吸附前Fe3O4@MnOx的SEM图 (b)吸附前Fe3O4@MnOx的TEM图,嵌入图为未负载MnOx的Fe3O4/C的TEM图 (c)未负载MnOx的Fe3O4/C的EDS图,嵌入图为吸附前Fe3O4@MnOx的EDS图

(d)吸附后Fe3O4@MnOx的SEM图 (e) 吸附后Fe3O4@MnOx的TEM图 (f) 吸附后Fe3O4@MnOx的EDS图
2.4.2 XRD表征
将Fe3O4@MnOx样品冷冻干燥后的粉末压片,进行XRD测试.从图5 (a)中可以看到样品Fe3O4@MnOx的2θ特征峰分别出现在35.46 °、62.9 °处,对应Fe3O4的(311)面和(440)面.而MnOx的特征峰不是特别明显,在2θ = 18 °~22 °范围内存在无定型的MnOx所产生的特征峰,结晶性较低[23-24].此外,从图5 (a)和5 (b)吸附前后的XRD衍射峰对比来看,结构仍相似,表明吸附过程中吸附剂的晶体结构未被破坏,但是吸附后的衍射峰变低了,这可能是U(VI)吸附在Fe3O4@MnOx上的缘故.
2.4.3 FT-IR表征
为了探究吸附剂的表面官能团,对Fe3O4@MnOx进行了FT-IR表征.FT-IR表征结果如图6所示.在510 cm-1和1 100 cm-1处的两个吸附峰对应于Mn-O表面的对称伸缩模式[25];于1645 cm-1处的吸收峰可能与吸附剂表面的C=O和HO-C=O基团有关,这是由外部热液碳层氧化引起的;在2362 cm-1处的峰由CO2控制[26].
此外,在3448 cm-1处出现了一个强而宽的振动峰,这与材料表面-OH和H2O的-OH有关,这不仅证明了Fe3O4@MnOx吸附剂上存在大量可以与铀离子结合的-OH吸附点,也表明该吸

附剂具有良好的亲水性和抗氧化性.

图6 Fe3O4@MnOx的FT-IR光谱
2.4.4 N2吸附脱附测试表征
通过N2吸附脱附测试来测定Fe3O4@MnOx的比表面积和孔径分布.结果如图7所示.

图7 Fe3O4@MnOx的N2吸附脱附等温线和孔径分布
从图中可以看到Fe3O4@MnOx的等温线剖面为典型的IV型且孔径大部分位于介孔区域,测得的比表面积为65.31 m2/g,平均孔径为12.36 nm,说明合成的Fe3O4@MnOx是一种介孔结构的吸附剂[27].
2.4.5 XPS表征
图8 (a)是Fe3O4@MnOx吸附U(VI)后的XPS全谱扫描图,该材料体系中主要有C、U、O、Mn、Fe五种元素.其特征峰分别在结合能为286.26 eV、383.26 eV、556.26 eV、655.26 eV、713.26 eV处.为进一步确定元素的存在形式,对元素Fe、Mn以及U进行分峰拟合,结果如图8(b)~8(d)所示.如图8 (b)所示,Fe3O4@MnOx材料中Mn元素的两个特征峰Mn 2p1/2和Mn 2p2/3的结合能分别为642.66 eV和654.26 eV,表明样品中的锰元素主要以Mn(IV)存在[28-29];在图8 (c)中,Fe元素由两个特征峰组成,其中Fe 2p3/2的结合能为711.76 eV,Fe 2p1/2的结合能为725.36 eV,这与Fe3O4的2p3/2和2p1/2特征峰相吻合,并且在715 eV~720 eV的范围内存在一个卫星特征伴峰,由此可确定Fe3O4@MnOx材料中Fe元素是以Fe3O4形式存在[30-31];如图8 (d)所示,U元素的特征峰U 4f7/2和U 4f5/2结合能分别为382.66 eV和393.41 eV,由此可以得出铀元素是以U(VI)存在的,这与我们的目标离子相吻合.

(a) Fe3O4@MnOx吸附U(VI) 后的XPS全谱扫描 (b) Mn 2p

(c) Fe 2p (d) U 4f的高分辨率光谱
3 结论
本文通过溶剂热法和模板法合成了新型Fe3O4@MnOx介孔球形材料,并将其用于U(VI)的吸附.通过间歇吸附实验结果表明,Fe3O4@MnOx对U(VI)的吸附行为更符合伪二级动力学模型和Langmuir等温模型,30 min内吸附几乎达到平衡.在温度为20 ℃,pH = 5时,其最大吸附量可达124 mg/g.表征结果表明,Fe3O4@MnOx具有明显的核壳式结构,Fe3O4被MnOx所包裹,有良好的多分散性和较规则的形状,且其孔径大小为介孔.因此,Fe3O4@MnOx对U(VI)具有良好的吸附性能,具有较高的实用价值和研究意义.
