神经网络的深度与宽度对药物分子pKa预测性能影响的研究
2021-05-08谢良旭薛亮亮李峰
谢良旭 薛亮亮 李峰

摘 要:pKa(解离常数)关系到药物分子在生物体内的吸收、代谢等过程。近年来,基于机器学习模型预测药物分子性质在药物筛选中获得广泛应用,神经网络可通过在深度与宽度两个方向上的扩展来增强模型的学习能力。以神经网络在药物分子pKa预测中的应用为例,比较了神经网络的深度与宽度对预测结果的影响。通过分析预测结果的均方差以及预测值与真实值之间的相关系数,系统地评估了模型的深度与宽度对预测性能的影响。基于定量的比较结果,提出了组合的神经网络模型计算方案。计算结果表明:深度神经网络模型在使用组合MACCS和ECFP指纹时,预测准确性超过了单一的宽度或深度神经网络。
关键词:人工智能;神经网络;深度学习;定量构效关系;药物发现;pKa
中图分类号:TP183 文献标识码:A 文章编号:2095-7394(2021)02-0001-08
pKa(解离常数)反映了药物分子重要的物理化学性质,是衡量药物分子在生物体内吸收、分布、代谢、毒性等药物代谢的重要指标。如何准确评估药物分子的pKa是药物设计领域需要解决的关键问题之一。生物体内的细胞都在稳定的pKa范围内活动,通过蛋白、磷酸盐、碳酸盐等提供适合的缓冲体系;因此,药物分子的pKa需要适用于所处的细胞环境。通过精准分析药物分子在不同环境中的pKa,可提前摒除不适合成药的分子,缩小药物筛选的范围。然而,通过实验测定分子的pKa费时费力,尤其是药物分子在体内环境中的pKa数值难以测定,而在理论计算方面,也尚未有成熟的开源计算软件[1];因此,亟需在研发过程中准确预测药物的pKa,从而有效降低药物研发的风险,控制药物研发成本。
pKa与药物分子结构相关,基于分子结构预测药物分子性质,是近几十年来生物信息学领域研究的热点。比如,早期的定量构效关系(Quantitative structure-activity relationship, QSAR)方法[2],使用数学模型建立分子的结构与物理化学性质以及生物活性之间的关系。QSAR是人类在药物发现领域使用最早的合理的药物设计方法,它不仅提供了一种预测药物分子性质的方法,而且为后续研究提供了重要的基本假设:药物分子的结构决定了该分子的物理、化学和生物等方面的信息,这些理化性质又决定了该分子在生物体内的生物活性[3]。从早期的回归分析、遗传算法,到现在热门的机器学习、深度学习等,基于分子结构预测其物理化学性质的假设,在药物设计等领域获得广泛采用[4-5]。近年来,人工智能方法成为药物设计领域中的热门技术,特别是深度学习方法兴起[6]。通过对数据集的训练,深度学习方法显著提高了预测结果的准确性和可靠性。人工智能方法提升了对分子溶解度、logD等物理性质的预测准确性[7-8];然而,对于药物分子pKa的计算,目前尚缺少系统的研究。
采用人工智能技术预测药物分子性质所面临的关键问题在于:如何将药物分子的分子结构转变为机器学习和深度学习可以直接识读和处理的文件格式?CHUANG K V[9]等人强调,有效的表征分子将直接影响到机器学习算法的准确性。而如何将药物分子的结构转变为数字形式,是连接药物化学与机器学习之间的桥梁。在化学信息学和生物信息学研究领域,一般采用分子描述符将分子结构编码为有用的化学信息。目前,应用较广泛的分子描述符是MACCS密钥[10]与拓展联通指纹ECFP[11]。MACCS密钥通过检索分子中是否存在字典中规定的子结构,将整个分子转变为二进制的化学信息。ECFP通过对每个原子周围的键连关系,搜索指定半径内的亚分子结构,并对所得到的亚分子分配一个数字符,以获得相同的编码序列,然后将数字符进行哈希化处理,从而得到一串特定长度的编码数字序列。ECFP密钥具有独一性,每个分子可编码为独特的数字串,因此也被称为ECFP指纹。MACCS密钥与ECFP指纹在之前的药物定量构效关系和机器学习中获得了广泛的应用,如用于药物分子相似性寻找、药物构效关系预测[12]以及对蛋白結合口袋的编码等[13]。
伴随着新的计算方法,深度神经网络在处理非线性的大数据挖掘问题中崭露头角[14-15]。深度神经网络具有广泛的应用性,尤其是随着计算机软硬件性能的提升,神经网络进入了高速发展的时期[16],目前所能处理的深度和宽度也得到了极大提升。当前研究的热点问题主要是神经网络在不同深度和宽度上的表现。一般而言,深度神经网络比浅层神经网络表现出更好的学习能力。图灵奖得主Yann LeCun提出,深度学习模型不能被简单的浅层模型所取代[17];而网络的深度对深度学习模型的性能起到了关键的作用。相比于深度神经网络,宽度神经网络由于没有多层连接,因此在网络层与层之间没有耦合,从而大大提高了网络计算速度[18]。增加神经网络的深度与宽度都能增加可学习参数的个数,进而可以增强神经网络对研究问题的拟合能力。然而,针对一些具体的问题,如:深度与宽度如何影响学习能力?如何选择合适的神经网络的深度与宽度?如何平衡网络深度与宽度的数目?目前仍未有系统性的评价。
本文以神经网络预测药物分子的pKa为例,通过对药物分子进行编码,系统比较神经网络的宽度与深度对预测结果准确性的影响,从而为神经网络对药物分子其他性质的预测提供指导。
1 材料和方法
1.1 数据集整理
选取药物分子数据库DrugBank进行数据整理。DrugBank数据库是对药物数据收集比较全面的数据库,目前收集了11 895种可能的药物分子[19]。利用脚本语言从数据库中抽取药物分子的结构和每个分子所对应的pKa数值,构建一个包含药物分子pKa的本地数据库。通过数据整理清洗发现:有8 656个药物分子包含pKa数值。为实现对神经网络的训练与验证,将输入数据按照8:2的比例分为训练集和测试集。将训练集中的数据用于超参数的优化过程,并在优化过程中随机选取20%的数据作为验证集;测试集中的分子是神经网络训练过程中未见过的分子,用于表征所训练的神经网络的泛化性能。
1.2 分子描述符
药物分子可以通过不同的化学式表示:分子式表示该化合物所包含的元素种类与含量;结构式表示该化合物的结构以及不同原子间的键连关系。MACCS密钥由166个描述符组成,每个描述符采用0或1来表示分子中是否包含相应的原子种类、成键信息、原子周围的环境等。ECFP因为包含了分子内的亚分子结构及联接关系,因此,在药物设计中发挥了独特的作用。本研究选取MACCS密钥与ECFP指纹对药物分子进行编码,并应用于药物分子的pKa预测中。采用RDKit软件将所选取的分子结构进行编码,对ECFP的指纹编码通过DeepChem软件实现,ECFP编码过程中的分子半径设置为2,哈希化为1 024个键值。如图1所示,以SIRT1的抑制剂分子为例,将药物分子结构转变为神经网络易于处理的分子描述符。1.3 所研究的神经网络架构
针对本次研究的数据预测和回归问题,通过神经网络预测药物分子的pKa。神经网络包含输入层、隐藏层和输出层,深度神经网络和宽度神经网络的架构如图2。输入层神经元的数目与输入数据的维度一致。宽度神经网络隐藏层的神经元数目分别为8、16、32、64、128、256、512、1 024、2 048和4 096。深度神经网络隐藏层的层数设置为1、2、4、8、16,神经元数目设置为32、64、128、256、512。神经网络的其他参数通过GridSearchCV超参数优化步骤获得,激活函数选择“relu”,优化器函数选择为“Adam”。在输出层之前使用dropout降低过拟合,根据参数优化结果将dropout的比例设置为0.5。
2 结果
2.1 宽度神经网络与神经元数目的关系
增加神经网络的深度与宽度都能增加模型的学习能力,那么,是否模型越宽、越深,其学习能力就越强呢?以预测药物分子的pKa为例,通过计算模型在不同宽度与深度情况下的拟合结果,能够分析模型的宽度与深度对结果的影响。
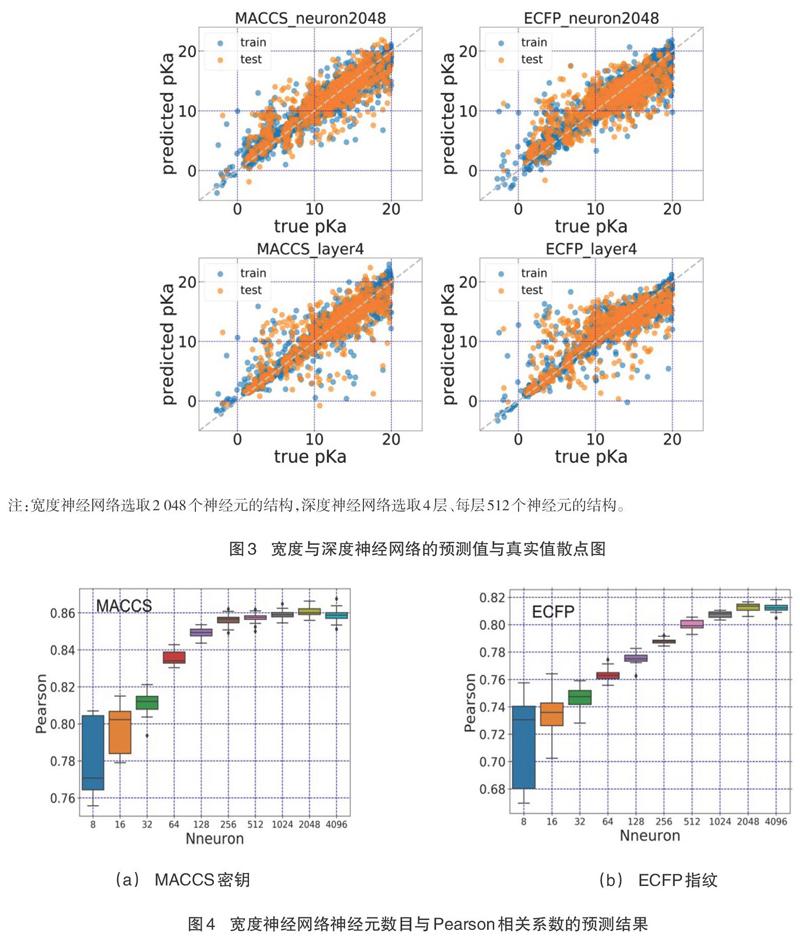
在使用MACCS密钥与ECFP指纹两种情况下,测试宽度神经网络的性能。如图3所示,为预测值与数据库中真实值的散点图。神经网络在测试集上的表现可通过计算神经网络预测值与数据库中真实值之间的Pearson相关系数定量比较,Pearson系数衡量了预测值与真实值之间的线性相关性。图4展示了使用不同的神经元数目时所计算的Pearson系数。可以发现:当增加宽度神经网络的神经元数目时,预测的结果逐渐达到平台值;当神经元的数目为2 048时,使用MACCS密钥与ECFP指纹所能达到的最佳拟合结果分别为0.864与0.816,即MACCS密钥能得到更好的拟合结果。MACCS密钥更容易达到模型学习能力的平台,当隐藏层神经元的数目为256时即达到了相对较好的预测结果;而在使用ECFP指纹时,需要隐藏层的神经元数目为2 048。MACCS密钥使用了166个键值;而ECFP使用了1 024个键值。由此可见,所需要的神经网络的宽度与输入的数据维度相关。为使宽度神经网络达到较好的预测能力,所使用的神经元数目要高于输入数据的维度。
2.2 深度神经网络与网络层数的关系
从以上对不同宽度神经网络的评测中发现,预测结果与所使用的神经元数目相关,当神经元数目少于32个时,预测结果的误差范围较广,预测结果的稳定性难以达到计算要求;因此,在衡量神经网络的层数对结果的影响时,分别选取神经元的数目为32、64、128、256和512。神经网络层数与预测能力的关系如图5所示。宽度神经网络和深度神经网络所使用的神经元的数目都不能低于32,否则会引起较大的预测误差。不同于宽度神经网络,深度神经网络的学习能力随着所使用的隐藏层的数目先升后降,在网络层数为4时,Pearson系数达到预测结果的峰值。在使用MACCS密钥与ECFP指纹时,最佳的预测结果为0.861和0.837。通常,加深网络的层数更容易带来优化问题,但由于深度网络中存在梯度不稳定的问题,因此,当使用的层数超过4时,预测结果反而出现了下降。可见,在进行特定任务的学习时,需要将网络使用的层数作为一个超参数进行优化。
2.3 神经网络宽度与深度对预测准确性影响的对比
进一步比较宽度与深度神经网络在训练过程的损失与预测结果的准确性。为更合理地比较预测结果,选取神经元数目为2 048时的宽度神经网络,与层数为4、神经元数目为512时的深度神经网络进行比较。两种网络使用了相同的神经元数目,不同神经网络中的损失函数结果如图6所示。
从损失函数上可以得出两个结论:(1)ECFP指纹比MACCS密钥容易带来过拟合的问题,ECFP指纹虽然在训练集上表现较好,但是在验证集上出现了较高误差。在预测药物分子pKa方面,MACCS密钥比ECFP指纹表现出更好的适用性。(2)深度神经网络在损失上的性能优于宽度神经网络,在训练集和验证集上的损失数值均低于宽度神经网络。
为比较预测结果的准确性,进一步计算预测结果与真实值之间的相对误差RMSE以及误差的统计分区,如表1。RMSE在0~0.5范围内的分类为“可接受区”(acceptable),在0.5~1.0范围内的分类为“可商榷区”(disputable),在大于1.0的分类为“不可接受区”(unacceptable)。分析RMSE和分类统计结果发现:深度神经网络在“可接受区”表现最佳,比例超过34%,說明深度神经网络比宽度神经网络获得了更好的准确性;虽然使用了相同的神经元数目,但是深度神经网络的表达能力远高于宽度神经网络;在使用2 048个神经元时,宽度神经网络的可组合形式为2 048个,而4层的深度神经网络的可组合形式为5124。因此,在预测药物分子pKa的实验中,深度神经网络的学习能力高于宽度神经网络,尤其是MACCS密钥在使用深度神经网络时获得最小的RMSE。
由此可見,网络的宽度和深度需要根据所研究的问题进行初步的训练与优化,神经元的数目或者所使用的网络层数与输入数据的尺度、所学习任务的复杂性相关。当使用的神经元宽度或者深度超过问题所需要的参数数目时,只会在训练中引入更多的噪声。
2.4 组合神经网络对预测性能的影响
通过增加单层神经网络的宽度或者网络的层数,可以获得更好的学习能力,从而提升神经网络的拟合能力;然而,扩大神经网络的规模会带来训练的问题,比如宽度神经网络出现了饱和,深度神经网络在网络层数较多时梯度不稳定。显然,不同模型各具优势,那么,能否使用组合的网络预测结果来提高模型的预测能力呢?借鉴集成学习的方法,可通过组合输入指纹或者组合网络模型的方式,获得多个差异化模型,并将不同模型的结果进行平均,从而完成对模型的组合计算。为此,本研究分别训练了MACCS与ECPF的组合指纹、深度与宽度的组合神经网络模型,通过模型融合策略中的平均法,分别计算4种组合情况下的Pearson系数。
计算结果如图7所示:4种组合所得到的Pearson系数均超过了相对应的单一神经网络模型的方法;MACCS密钥和ECFP指纹在使用4层神经网络、每层512个神经元网络架构时的预测结果优于其他3种组合方式;两种不同的指纹输入方式所训练出的网络模型能较好地改善泛化误差,尤其是MACCS密钥与ECFP指纹在深度神经网络组合的情况下,Pearson系数为0.88,达到了最佳的计算结果。可见,在使用相同的神经元数目时,两种模型的组合获得了比单独使用宽度或深度神经网络更好的预测能力。
组合模型方法因其具有高度的灵活性和较强的适用性,近年来得到了广泛的应用。研究结果表明:组合模型方法可以抵消不同网络模型的方差,有利于提高模型的预测准确性,并且,组合模型的计算结果超过了任意单一的模型。Gao Y等人[20]在近期的综述中,进一步分析了组合模型方法的应用领域以及存在的机遇与挑战,为组合模型在生物信息领域的发展指明了方向。在今后的研究中,将进一步深入地组合神经网络模型以实现模型的互补,消除计算结果的偏差,从而获得更好的预测结果。
3 结语
本研究探讨了神经网络不同的宽度和深度对预测药物分子pKa的影响。通过系统地计算比较,发现在一定范围内增加神经网络的宽度或者网络的层数可以获得更好的学习能力;在使用相同神经元数目时,深度神经网络可以获得更高的准确性。然而,宽度神经网络模型会随着宽度增加而出现饱和,深度神经网络存在最适网络层数问题;因此,为进一步提升预测结果,提出了一种通过平均法组合使用MACCS密钥与ECFP指纹的深度神经网络计算方法,使得预测的Pearson系数达到0.88,超过了单一神经网络、单一分子描述符的预测能力。本研究提出的神经网络的宽度、深度以及组合策略,也为将来进一步应用神经网络预测药物的其他性质提供了有益的尝试。
参考文献:
[1] MANSOURI K, CARIELLO N F, KOROTCOV A,et al. Open-source QSAR models for pKa prediction using multiple machine learning approaches[J]. Journal of Cheminformatics,2019,11(1):60.
[2] DANISHUDDIN, KHAN A U. Descriptors and their selection methods in QSAR analysis: paradigm for drug design [J]. Drug Discov Today,2016,21(8):1291-1302.
[3] CHERKASOV A, MURATOV E N, FOURCHES D, et al. QSAR modeling: where have you been? where are you going to?[J]. Journal of Medicinal Chemistry,2014,57(12):4977-5010.
[4] WENZEL J, MATTER H, SCHMIDT F. Predictive multitask deep neural network models for ADME-Tox properties: Learning from large data sets[J]. Journal of Chemical Information and Modeling,2019,59(3):1253-1268.
[5] MIYAO T,KANEKO H,FUNATSU K. Inverse QSPR/ QSAR analysis for chemical structure generation (from Y to X)[J]. Journal of Chemical Information and Modeling,2016,56(2):286.
[6] JING Y, BIAN Y, HU Z, et al. Deep learning for drug design: an artificial intelligence paradigm for drug discovery in the big data era[J]. AAPS Journal,2018,20(3):58.
[7] WU Z, RAMSUNDAR B, FEINBERG EVAN N,et al. MoleculeNet: a benchmark for molecul AR machine learning [J]. Chemical Science,2018,9(2):513-530.
[8] FU L,LIU L,YANG Z J,et al. Systematic modeling of log D(7.4) based on ensemble machine learning, group contribution, and matched molecular pair analysis[J]. Journal of Chemical Information and Modeling,2020,60(1):63-76.
[9] CHUANG K V, GUNSALUS L M, KEISER M J. Learning molecular representations for medicinal chemistry[J]. Journal of Medicinal Chemistry,2020.DOI:10.1021/acs.jmedchem.0c00385.
[10] DUAN J, DIXON S L, LOWRIE J F,et al. Analysis and comparison of 2D fingerprints: insights into database screening performance using eight fingerprint methods[J]. Journal of Molecular Graphics & Modelling,2010,29(2):157-170.
[11] ROGERS D,HAHN M. Extended-connectivity fingerprints[J]. Journal of Chemical Information and Modeling,2010,50(5):742-754.
[12] PRASAD S, BROOKS B R. A deep learning approach for the blind LogP prediction in SAMPL6 challenge[J]. Journal ofComputer-aided Molecular Design,2020,34(5):535-542.
[13] LI L,KOH C C,REKER D,et al. Predicting protein-ligand interactions based on bow-pharmacological space and Bayesian additive regression trees[J]. Scientific Reports ,2019,9(1):7703.
[14] XU Y,YAO H,LIN K. An overview of neural networks for drug discovery and the inputs used[J]. Expert Opin Drug Discovery,2018,13:1091.
[15] 孫志远,鲁成祥,史忠植,等. 深度学习研究与进展[J]. 计算机科学,2016,43(2): 1-8.
[16] CHEN H, ENGKVIST O,WANG Y,et al. The rise of deep learning in drug discovery[J]. Drug Discovery Today,2018,23(6):1241-1250.
[17] BENGIO Y,LECUN Y. Scaling learning algorithms towards AI[C]// Large-Scale Kernel Machines,2007.
[18] CHEN C L P, LIU Z. Broad learning system: an effective and efficient incremental learning system without the need for deep architecture[J]. IEEE Transactions on Neural Networks and Learning Systems, 2018,29(1):10-24.
[19] WISHART D S, FEUNANG Y D, GUO A C,et al. DrugBank 5.0: a major update to the DrugBank database for 2018[J]. Nucleic Acids Research,2018,46(D1):1074-1082.
[20] CAO Y,GEDDES T A,YANG J Y H,et al. Ensemble deep learning in bioinformatics[J]. Nature Machine Intelligence,2020,2(9): 500-508.
责任编辑 盛 艳
