溶剂热法合成Fe3O4纳米微粒及其调控机制*
2021-05-08李永贵吴依琳宋晓蕾麻文效
李永贵,吴依琳,宋晓蕾,麻文效
(1.内蒙古工业大学 轻工与纺织学院,呼和浩特 010080;2.闽江学院 福建省新型功能性纺织纤维及材料重点实验室,福州 350108;3.闽江学院 服装与艺术工程学院,福州 350108)
0 引 言
Fe3O4作为一种多功能磁性材料,除了具有高饱和磁化强度、高磁导率和低损耗等特性,还具有优良的耐光性、耐热性、耐腐蚀性等,使其在吸附、催化、磁介质、生物医学等领域展示出极高的应用价值[1-5],因此,Fe3O4一直是磁性材料领域研究的热点。制备Fe3O4纳米微粒的方法有许多种,如溶剂热法、共沉淀法、微乳液法和溶胶凝胶法等[6-10]。其中,溶剂热法是在高温高压密闭条件下进行的,该方法不仅能够促进反应的进行,制备出常温常压条件下无法制备的材料,还能有效避免组分的挥发[11]。目前,国内外已有许多研究者采用溶剂热法制备Fe3O4纳米微粒,但还未见比较系统的关于其最优合成条件的相关报道。为制备出磁性强、形貌尺寸均一且分散性好的Fe3O4纳米微粒,本文采用溶剂热法,通过分析其反应原理,调控反应时间、反应温度和PEG用量,分析并优化溶剂热法的反应工艺,为制备Fe3O4纳米微粒提供参考。
1 实 验
1.1 试 剂
六水合三氯化铁(FeCl3·6H2O)、乙二醇((CH2OH)2,EG),天津市恒兴化学试剂制造有限公司;无水乙酸钠(CH3COONa),西陇科学股份有限公司;聚乙二醇(PEG,分子量2000),上海麦克林生化有限公司。所有试剂均为分析纯。
1.2 实验方法
溶剂热法制备Fe3O4纳米微粒方法如下。取0.01 mol FeCl3·6H2O加入40 mL EG溶液中,并加入一定量PEG,通过磁力搅拌至完全溶解。再取0.1 mol CH3COONa 加入20 mL EG溶液中,搅拌至完全溶解,并缓慢滴加至铁盐溶液,得到浓稠的褐色溶液。随后,将其转移并密封至100 mL高压反应釜中,在高温烘箱中反应。反应结束后,冷却到室温,用EG溶液与去离子水轮流洗涤产物数次。最后,通过磁铁收集产物,将其烘干即得到Fe3O4纳米微粒。为探究Fe3O4的合成过程,设置多组反应时间(A)、反应温度(B)和PEG的用量(C)参数进行试验,详细数据如表1 所示。
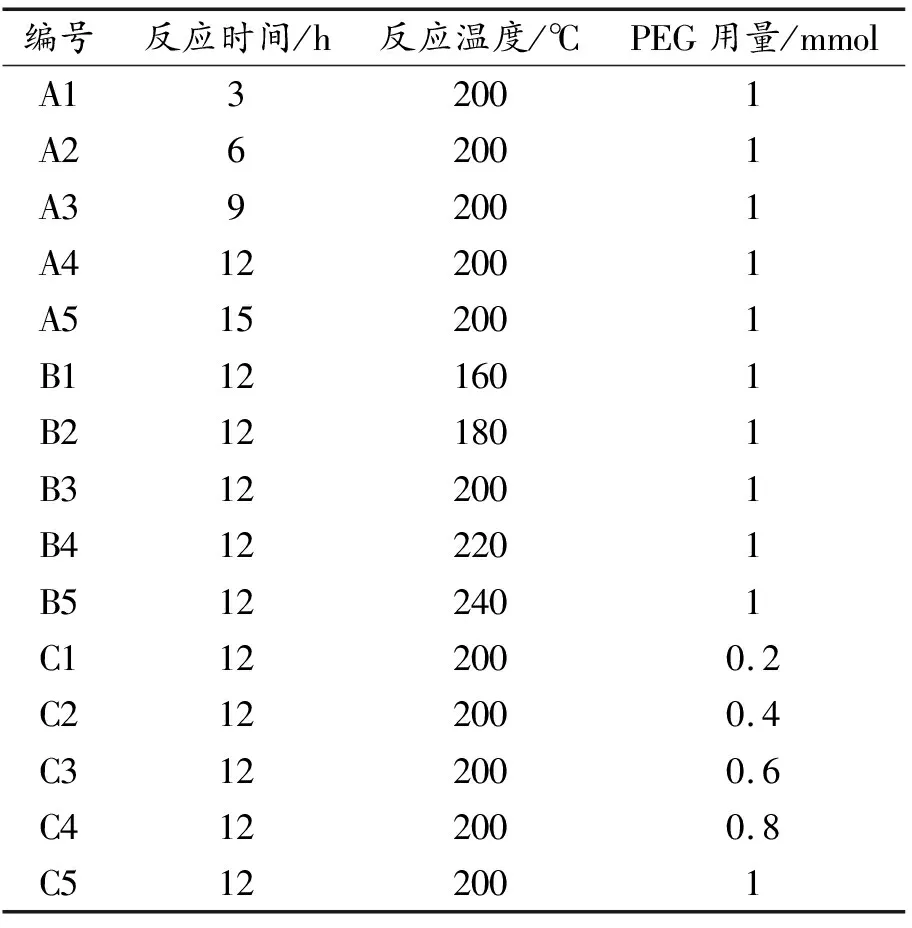
表1 实验设计参数表
1.3 表征与测试
采用S4800型场发射扫描电子显微镜(SEM,日本日立公司)观察产物的表面形貌;采用D8 ADVANCE 多晶(粉末)X射线衍射仪(XRD,德国布鲁克公司)对产物的晶体结构进行表征;采用IS50傅里叶变换红外光谱仪(FT-IR,美国赛默飞世尔公司)分析产物的特征官能团;采用PPMS-9振动样品磁强计(VSM,美国量子设计公司)测试产物的磁性能。
2 结果与讨论
2.1 合成机理分析
在整个合成过程中,EG作为介质溶液,为反应提供温和的环境。同时,其在高温条件下也可作为还原剂,将部分Fe3+还原成Fe2+;CH3COONa提供的CH3COO-与FeCl3·6H2O中的微量水分发生水解反应,生成OH-,为反应提供弱碱性环境。最后,Fe2+与Fe3+在高温高压的条件下与OH-反应生成Fe3O4纳米微粒。采用PEG作为表面活性剂,目的是提高纳米微粒的分散性。实验体系中存在的反应方程式如式(1, 2)所示。
(1)

(2)
2.2 反应时间对产物的影响机理
图1显示了在不同反应时间的条件下产物的表面形貌变化。可以明显看出,当反应时间为3 h(图A1),已生成部分球形微粒,同时存在许多不规则块状物质,这是由于反应时间较短导致反应不彻底,还未完全生成Fe3O4纳米微粒反应就被终止。当反应时间为6 h(图A2),已生成大量Fe3O4纳米微粒,但球与球之间存在粘连现象,且有许多小球团聚严重,说明反应时间还不够,未能反应充分。当反应时间为9 h(图A3),已完全生成Fe3O4纳米微粒,但其尺寸不均一,仍存在许多粒径较小的球体,说明此时反应已完全进行,但晶体未得到充分生长。当反应时间为12 h(图A4),生成的Fe3O4纳米微粒尺寸均一(主体粒径在400 nm左右),形态稳定,表面比较光滑,仅有少量粒径较小的晶粒存在,此时晶体已充分生长。当反应时间为15 h(图A5),许多Fe3O4纳米微粒的表面开始出现凹痕,甚至破裂,说明晶体过度生长,破坏了纳米微粒的结构,也导致了粒径分布不均,同时又出现了许多小的晶粒,其形态萎缩。这是因为产物经历了Ostwald熟化过程,即大晶体的生长以牺牲小晶体为代价,当大晶体继续生长,小晶体会出现萎缩甚至是消溶的现象[12]。因此,反应时间为12 h最为合适。

图1 不同反应时间下产物的SEM图:(A1)3 h;(A2)6 h;(A3)9 h;(A4)12 h;(A5)15 h
不同反应时间下产物的FT-IR图谱如图2所示。由图可见,产物A2/A3/A4/A5对应的曲线均在579 cm-1处出现了明显的特征峰,为Fe—O伸缩振动所致,表明产物确实是Fe3O4。但是,产物A1的曲线并没有出现明显的Fe—O伸缩振动峰,结合A1的SEM图,可以得知产物为部分Fe3O4纳米微粒和大量块状物质,Fe—O特征峰很可能是被500~700 cm-1处出现的更强的吸收峰所覆盖。此外,在3 455 cm-1处出现了宽吸收峰,为水分子中—OH的伸缩振动峰。在1 651 cm-1处出现了CO伸缩振动峰,在1 476 cm-1处出现了—CH3面内弯曲振动峰,均可能是因为在3 h条件下乙酸钠反应不完全所致。在1 130 cm-1处出现了C—O伸缩振动峰,在683 cm-1处出现了C—H面外弯曲振动峰,为乙二醇的特征基团。由此可知,反应时间为3 h不能完全生成球形Fe3O4纳米微粒。
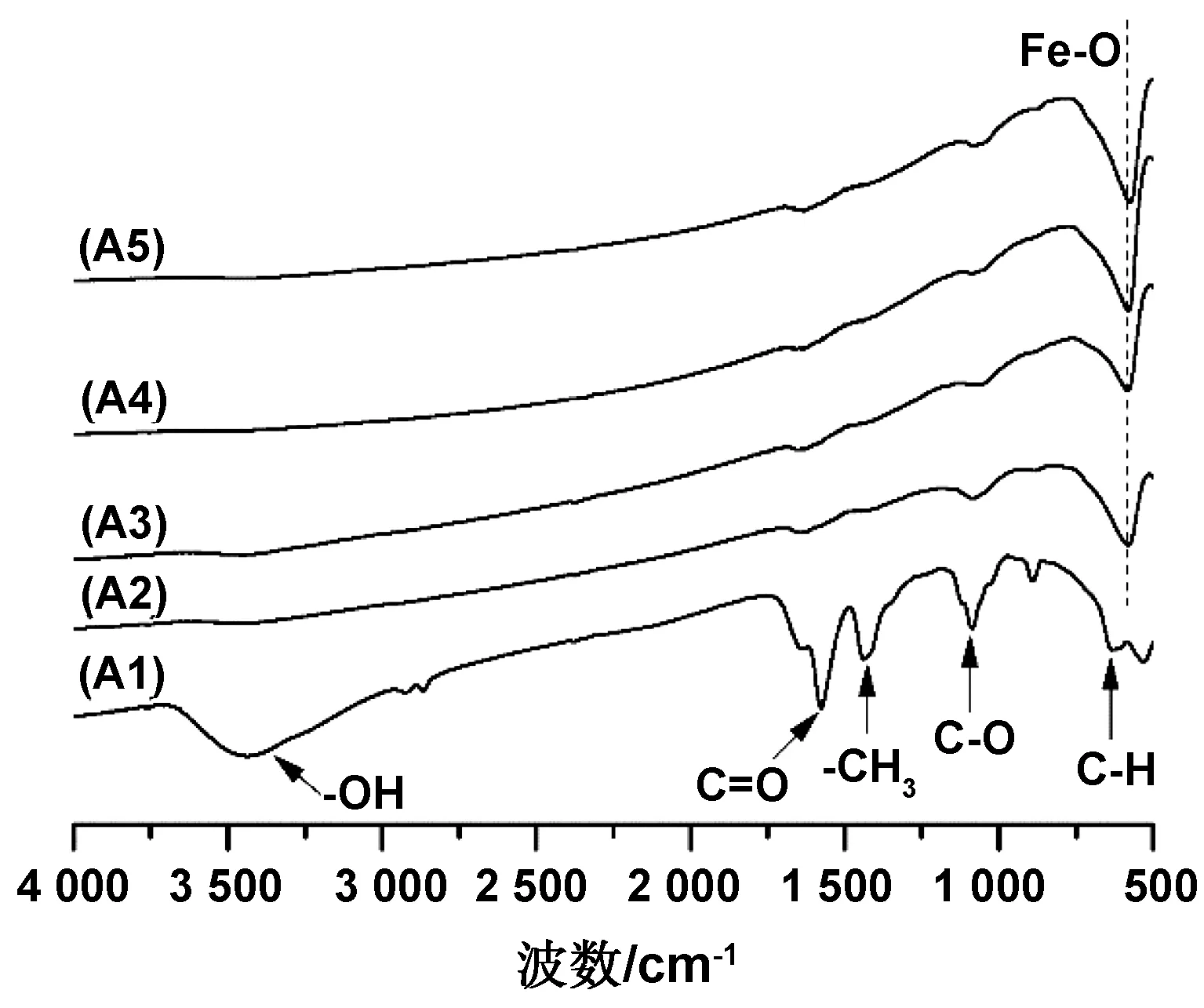
图2 不同反应时间下产物的FT-IR图谱
图3为不同反应时间下产物的XRD图谱。如图所示,曲线A1在衍射角为25.2°和31.3°处分别出现了(022)和(-223)衍射峰,该峰位置及强度对应于CH3COONa标准卡片(JCPDS No.29-1160),这可能是因为在反应时间为3 h的条件下,反应未完全进行,导致反应物CH3COONa仍有残留。此外,当衍射角为30.2°、35.6°、43.1°、53.5°、57.1°、和62.5°时,分别出现了强度非常微弱的特征衍射峰(220)、(311)、(400)、(422)、(511)和(440),其衍射峰位置与强度与Fe3O4标准卡片(JCPDS No19-0629)基本相符,这说明反应进行了一半,已生成部分Fe3O4纳米微粒。曲线A2/A3/A4/A5也在相同的位置出现了属于Fe3O4纳米微粒的特征衍射峰,无其他杂峰,说明在6/9/12/15 h 条件下产物为纯Fe3O4纳米粒子,但是,随着反应时间的增加,衍射峰强度也增加;峰强度越大,代表产物结晶度越高[13]。
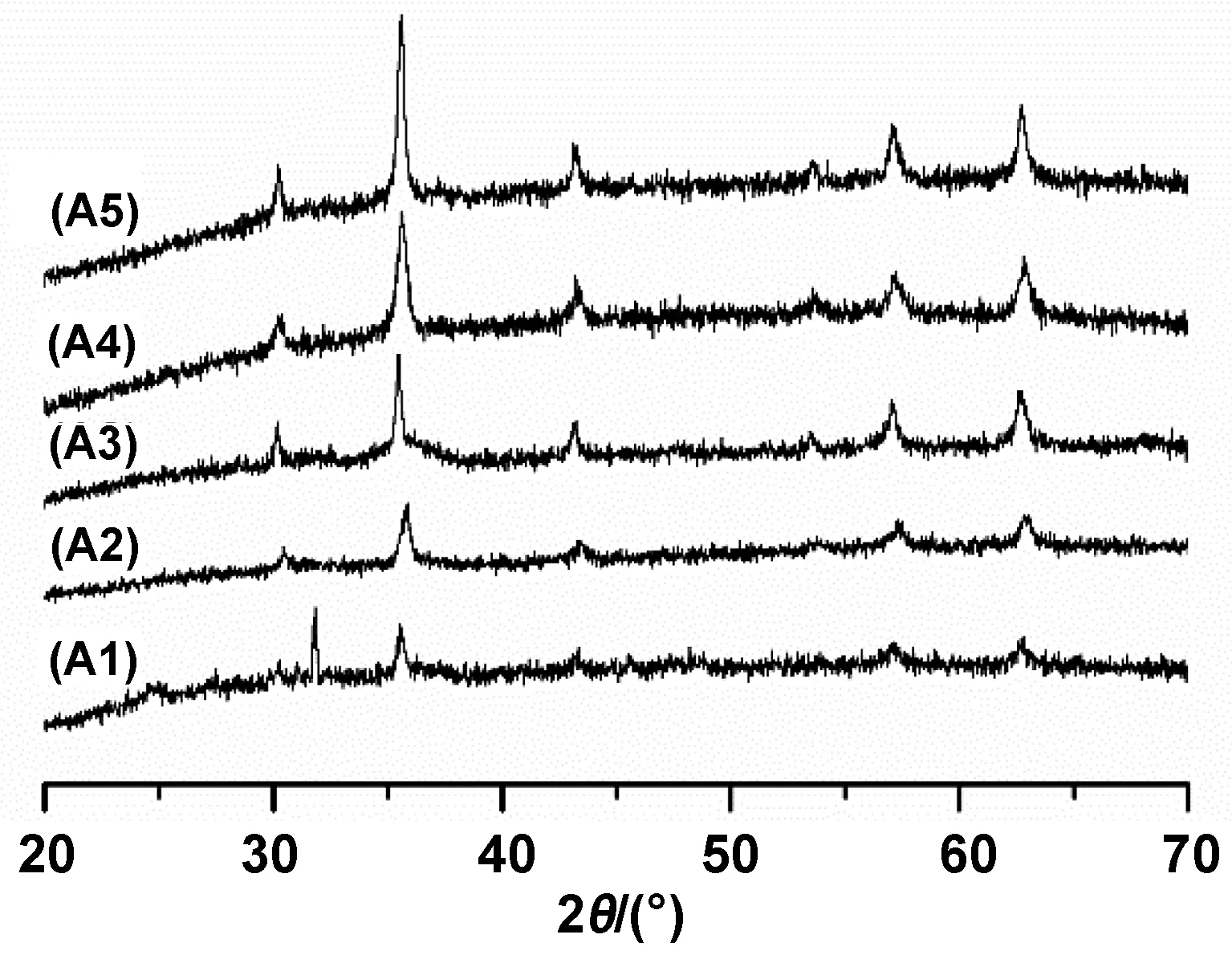
图3 不同反应时间下产物的XRD图谱
2.3 反应温度对产物的影响机理
图4为不同反应温度下产物的SEM图。可以观察到,当反应温度为160 ℃(图B1),产物为不规则块状物质,未出现任何球形微粒,这说明在该温度条件下,不能生成Fe3O4纳米微粒。当反应温度为180 ℃(图B2),产物为大量的球形Fe3O4纳米微粒,同时存在少量块状物质,且部分球形微粒有团聚现象,这表明该温度条件足以让反应正常进行,但不足以让晶体充分生长。当反应温度为200 ℃(图B3),生成的产物均为表面光滑的球形结构Fe3O4纳米微粒,其形态规整,大小均一,平均粒径约为400 nm,此时的反应温度刚好超过溶剂(EG)的沸点,使得反应体系压力增大,有利于晶体的生长[14]。当反应温度为220 ℃(图B4),可以观察到,少量Fe3O4纳米微粒的表面开始变得粗糙、出现凹陷。当反应温度为240 ℃(图B5),大量晶体表面都是粗糙且有凹陷的,甚至有少量晶体破裂,这是因为温度过高,导致晶体过度生长。由此认为,200 ℃为比较合适的反应温度。

图4 不同反应温度下产物的SEM图:(B1)160 ℃;(B2)180 ℃;(B3)200 ℃;(B4)220 ℃;(B5)240 ℃
不同反应温度下产物的FT-IR图谱如图5所示。由图可见,产物B2/B3/B4/B5对应的曲线均在579 cm-1处出现了Fe-O伸缩振动峰,说明在180/200/220/240 ℃条件下均可合成Fe3O4纳米微粒。而产物B1对应的曲线与2.1中产物A1的曲线在同样的位置出现了更强的特征吸收峰,且对应的基团也一致,这说明160 ℃的温度条件不能合成Fe3O4纳米微粒。
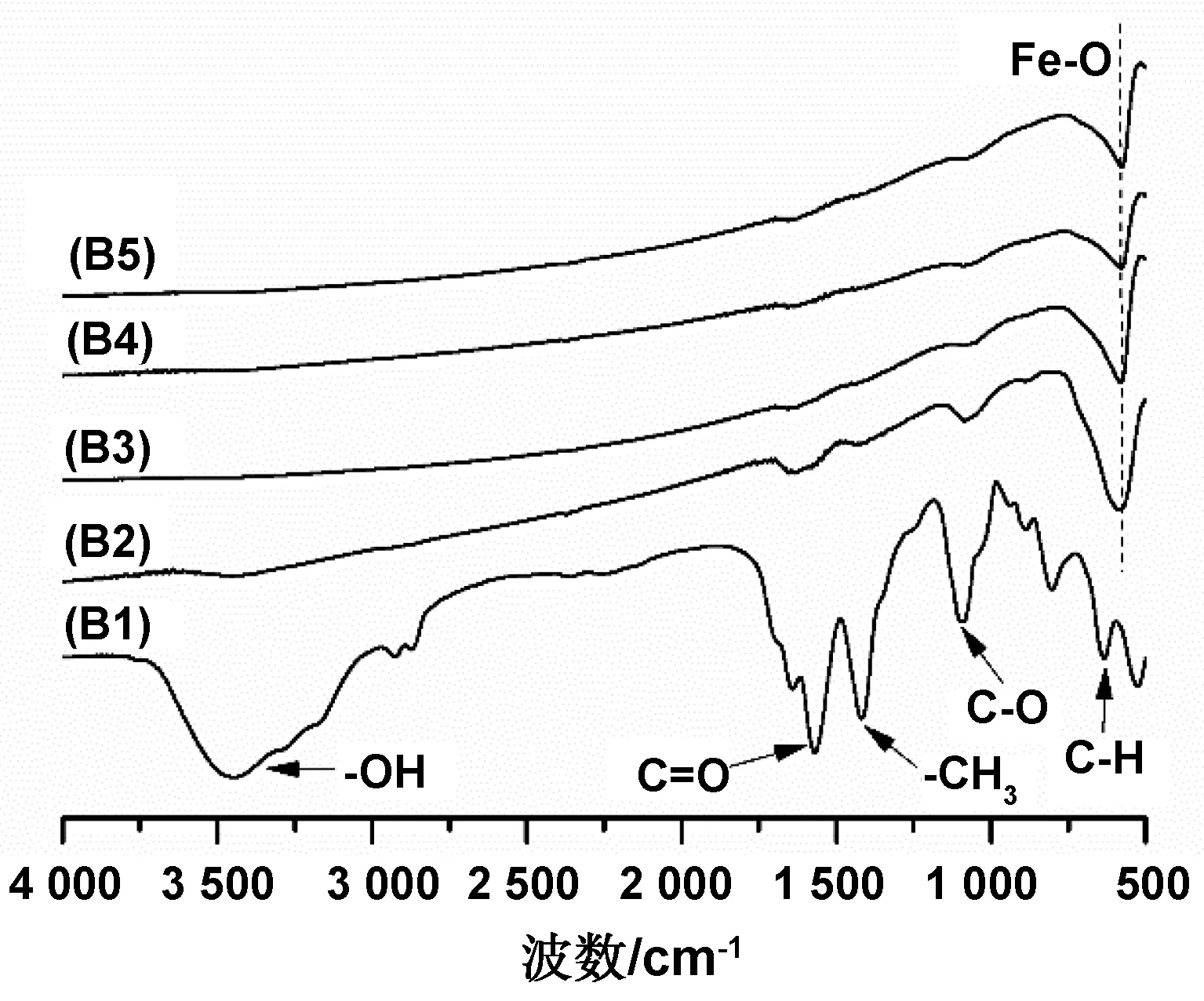
图5 不同反应温度下产物的FT-IR图谱
图6为不同反应时间下产物的XRD图谱。如图所示,曲线B1在衍射角为25.2°、29.7°、31.3°和36.7°处分别出现了(022)、(-402)、(-223)和(-133)衍射峰,根据标准卡片(JCPDS No.29-1160),确定该峰属于CH3COONa,此外未出现其他的衍射峰,这表明当反应温度为160 ℃,不足以让反应物相互反应生成Fe3O4。而当温度为180/200/220/240 ℃时,其对应的曲线B2/B3/B4/B5均在衍射角为30.2°、35.6°、43.1°、53.5°、57.1°、和62.5°处,分别出现了特征衍射峰(220)、(311)、(400)、(422)、(511)和(440),其位置与强度对应于Fe3O4标准卡片(JCPDS No19-0629),这说明当温度为180/200/220/240 ℃时,均能够成功制备Fe3O4纳米微粒。
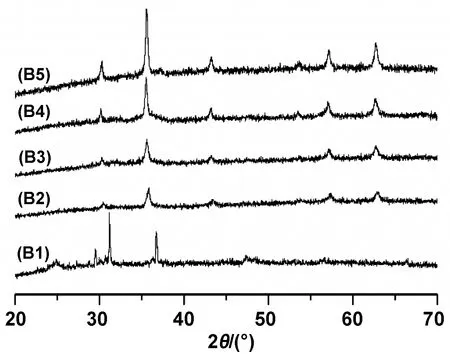
图6 不同反应温度下产物的XRD图谱
2.4 PEG用量对产物的影响机理
图7主要呈现了不同PEG用量对产物分散性的影响。当PEG用量为0.2/0.4/0.6 mmol(图C1/C2/C3),Fe3O4纳米微粒均呈现大面积的团聚现象,随着PEG用量的增加,团聚现象有所减少,这是由于PEG作为分散剂,能够产生空间位置阻隔的作用,阻碍颗粒之间相互接触,由此提高产物的分散性。当PEG用量为0.8 mmol(图C4),粒子整体存在小面积的团聚,但相比于样品C1,分散性有了较大的提高。当PEG用量为1 mmol(图C5),可以观察到Fe3O4纳米微粒已无团聚现象,每个粒子的轮廓清晰可见,分散性得到了明显的改善。此外还可以发现,当PEG用量从0.2 mmol增大到0.6 mmol时,Fe3O4纳米微粒的粒径也随之增大。当PEG用量超过0.6 mmol时,Fe3O4纳米微粒的整体粒径已趋于稳定,不再随着PEG用量的增加而增大,这是因为PEG作为表面活性剂,有利于晶体生长,但是,当其用量超过一定的浓度,就会开始抑制晶体的生长[15-16]。

图7 不同PEG用量下产物的SEM图:(C1)0.2 mmol;(C2)0.4 mmol;(C3)0.6 mmol;(C4)0.8 mmol;(C5)1 mmol
不同PEG用量下产物的FT-IR图谱如图8所示。由图可见,产物C1/C2/C3/C4/C5对应的曲线在579 cm-1处均出现了Fe-O伸缩振动峰,且未在其它位置出现明显的特征吸收峰,这是因为PEG在反应过程中是作为分散剂,其用量的多少主要是影响产物的分散性,不会改变产物的成分结构。
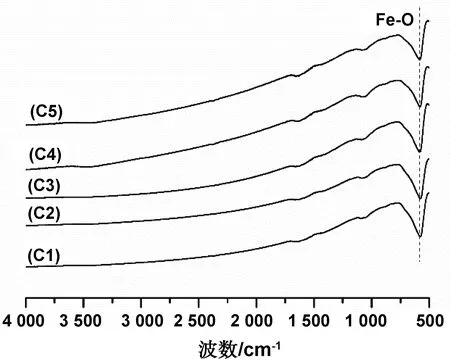
图8 不同PEG用量下产物的FT-IR图谱
图9为不同PEG用量下产物的XRD图谱。如图所示,曲线C1/C2/C3/C4/C5均在衍射角为30.2°、35.6°、43.1°、53.5°、57.1°、和62.5°处,分别出现了特征衍射峰(220)、(311)、(400)、(422)、(511)和(440),其位置与强度对应于Fe3O4标准卡片(JCPDS No19-0629)。随着PEG用量从0.2 mmol增加到0.6 mmol,曲线C1/C2/C3的衍射峰强度逐渐增大,这是因为PEG作为表面活性剂会吸附到晶粒表面,并降低比表面能,促使晶粒逐渐长大,结晶性逐渐完善。而PEG用量超过0.6 mmol时,衍射峰强度没有继续增大(C3/C4/C5的衍射峰强度几乎一致),这是因为PEG用量增大,晶粒表面对其吸附量也增大,且在晶体表面会形成有序的覆盖层,使得晶体生长速率变慢[17-18]。因此,产物C3/C4/C5的粒径大小相似,结晶度也几乎一致。
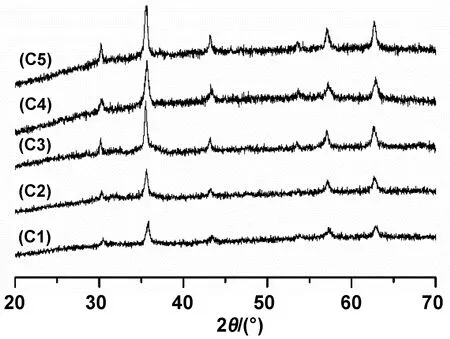
图9 不同PEG用量下产物的XRD图谱
2.5 Fe3O4纳米微粒的磁性能分析
通过对反应时间、反应温度和PEG用量的调控,最终得知12 h、200 ℃和1 mmol为最优合成条件,选取该反应条件下的产物测试磁性能,其在室温下的磁滞回线如图10所示。由图可知,曲线闭合,呈典型的“S”型,Fe3O4纳米微粒的饱和磁化强度(Ms)较高,为68.91 A·m2/kg,对磁铁感应及其灵敏,在合成阶段完全可以用磁铁收集产物。另外,Fe3O4纳米微粒的矫顽力(Hc)和剩余磁感应强度(Mr)如图中左上角的放大图所示,分别为1954.97 A/m 和2.33 A·m2/kg,这两个值几乎小到可以忽略不计,这些特征都与软磁材料的特点相符,这说明合成的Fe3O4纳米微粒具有良好的磁性能,可用于磁性领域相关研究及应用[19-20]。
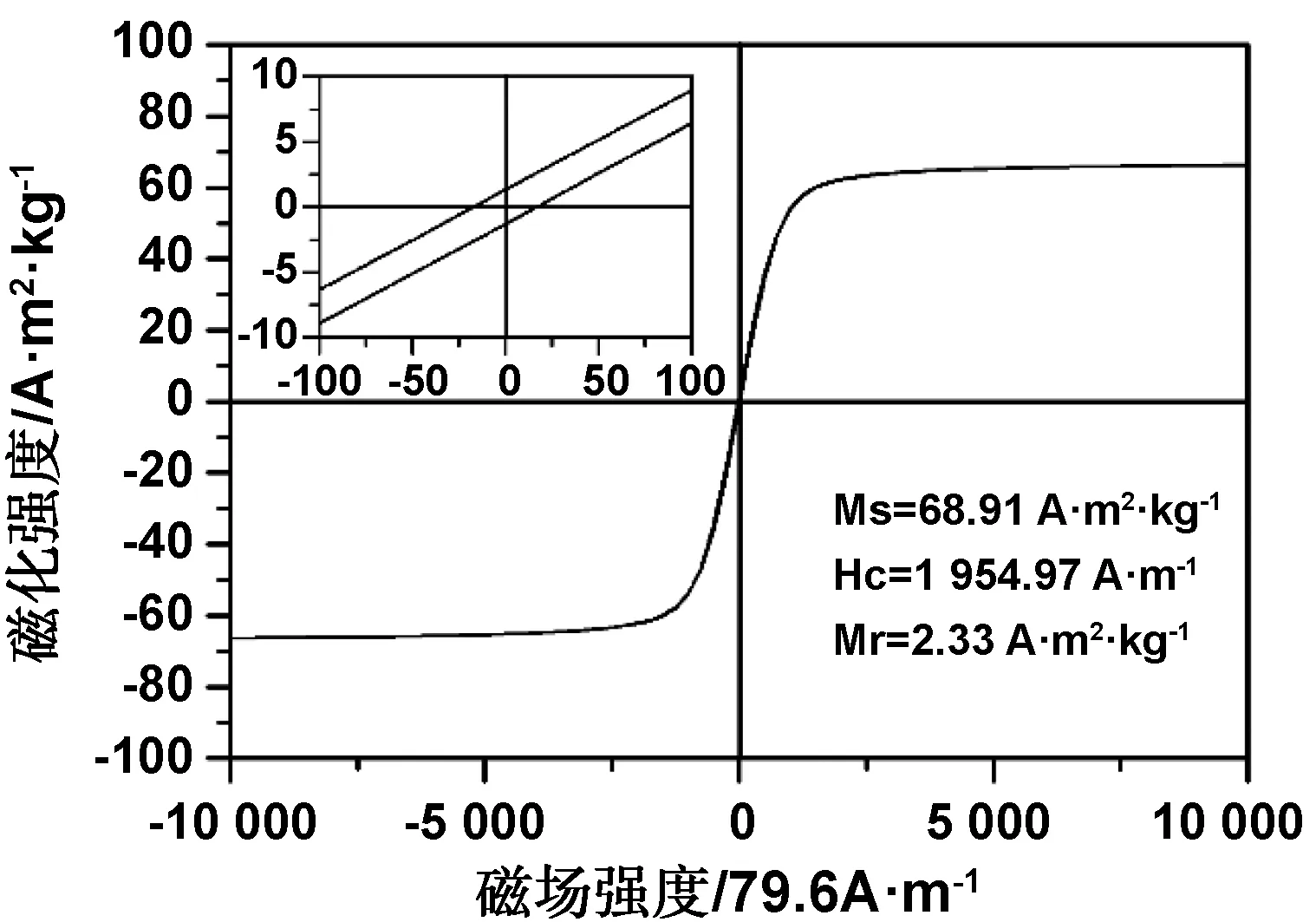
图10 Fe3O4纳米微粒的磁滞回线图
3 结 论
(1)采用溶剂热法合成Fe3O4纳米微粒并调控其反应条件,反应时间12 h、反应温度200℃、PEG用量1 mmol为最优合成条件。
(2)在最优合成条件下制备的Fe3O4纳米微粒为尺寸均一、形态稳定的球体,其平均粒径约为400 nm,粒子分散性好,未出现团聚现象;该Fe3O4纳米微粒具有良好的磁性能。
