双黄连色谱指纹图谱和谱效关系研究进展*
2021-05-07杜怡婷李冬梅
李 楠,杜怡婷,马 鑫,李冬梅
(华北理工大学药学院,河北唐山 063210)
0 引 言
中药是中华民族的瑰宝,是中国特色和文化自信的组成部分.中药品质是中药临床用药安全有效的基础,亦是保障中药产业可持续发展的生命线,直接关乎中药标准化和国际化发展.中药质量控制具有一定难度,这是因为中药多具以复方制剂和多成分给药的特点,即“多组分、多靶点和多通路”[1],其质量标准也从过去的定性鉴别为主,发展到同时测定多成分、多靶点的整体质量评价.近年来,一测多评法、中药指纹图谱技术、快速检测技术和仿生技术等多种现代分析方法和技术广泛应用于中药质量控制研究[2],使其朝着标准化、现代化和全面化的方向发展.
中药色谱指纹图谱技术指中药经适当前处理后,采用色谱分析方法,得到能够标示其化学特征的色谱图[3].该技术能够很好地反映中药品质的真实性、一致性和稳定性,得到国内外学者的普遍认可.为进一步将中药所含化学成分和其药效相关联,建立能够通过色谱指纹图谱而预测其药效的质量控制和评价新模式[4],谱效关系的研究应运而生[5-6].有关中药谱效关系的研究论文不多,柯昌虎等[7]从色谱指纹图谱、光谱指纹图谱和联用技术指纹图谱等方面对金银花指纹图谱的研究方法进行归纳总结,为制定科学可靠的金银花质量评价体系提供依据;王常瞵等[8]对中药黄芩色谱指纹图谱、光谱指纹图谱和生物指纹图谱的研究进展进行综述,为合理评价和控制黄芩药材品质提供依据.
双黄连是一味由金银花、黄芩和连翘组成的中成药,具有疏风解表、清热解毒的功效,作为临床常用药,具有广泛的抗菌、消炎和解毒作用,如抑制肺炎双球菌,治疗小儿支原体肺炎、小儿疱疹性咽峡炎,治疗口腔溃疡甚至有抗结肠癌的活性等[9].目前,尚未见双黄连指纹图谱和谱效关系方面的综述报道.为了建立全面、系统的双黄连质量控制和评价体系,以指导其临床用药.本文从不同剂型双黄连样品的前处理方法,不同色谱分析方法对其色谱指纹图谱的研究方法进行归纳整理,分析目前双黄连谱效关系研究进展,并对其发展前景进行展望.
1 双黄连样品前处理
临床常用双黄连剂型有口服液、注射剂、片剂、颗粒剂、胶囊剂和凝胶剂等,不同剂型的样品前处理方法略有不同.
1.1 口服液
双黄连口服液型样品前处理方法主要是通过溶剂对样品进行稀释或稀释、过滤后备用.如陈俊[10]将400μL双黄连口服液置于25 mL的容量瓶中并加入体积分数为50%(文中如无特殊说明时均为体积分数)甲醇溶剂,超声20 min后室温放置,再次加入50%甲醇定容后摇匀,以此作为供试品备用.冯宏玲等[11]取10支双黄连口服液混合,量取500μL混合液置于10 mL容量瓶中,同样加入50%甲醇进行定容,摇匀后用0.45 μm微孔滤膜进行滤过;王新宏等[12]取双黄连口服液5支,混匀后量取500μL用50%甲醇定容至10 mL,作为供试品溶液备用.
1.2 注射剂
双黄连冻干粉类注射剂型通常需要经溶剂溶解,之后前处理方法类似于口服液的处理方法.如郭洁等[13]选择双黄连冻干粉制备供试品溶液,取适量样品至10 mL容量瓶,用30%甲醇定容,超声分散备用;Luan等[14]直接将双黄连注射液经 0.45 μm尼龙膜过滤后,进行样品分析;Zhang等[15]首先用50%甲醇稀释双黄连注射液,再以0.22 μm滤膜进行滤过后备用.
1.3 片剂
双黄连片剂型样品前处理方法是通过对样品研磨、超声溶解和过滤后备用.如唐佩琴[16]取双黄连片剂研细后,称质量为1.0 g,置于50 mL棕色容量瓶中并加入60%甲醇,恒温超声45 min后,待样品温度降至室温,继续加60%甲醇定容并摇匀、过滤后备用;赵洪涛等[17]取20片双黄连片剂于研钵中研细,称取粉末0.2 g,用50%甲醇/水溶解,超声30 min后,再用50%甲醇/水补齐溶液至10 mL,0.45 μm滤膜过滤,得到待分析供试品溶液.
1.4 颗粒剂
双黄连颗粒剂型样品前处理方法类同于片剂.如周志等[18]取3袋双黄连颗粒的内容物,研细后,称取1.0 g,置具塞锥形瓶中,加50%甲醇并超声处理20 min,最后用 0.45 μm滤膜过滤备用;高利利等[19]也是将双黄连颗粒研细后,采用溶剂溶解,过滤除杂等,制得待测样品.
1.5 胶囊剂
双黄连胶囊剂型样品在分析时取胶囊内的内容物,后续样品处理方法与片剂和颗粒剂类似,也可采用溶剂回流提取的样品前处理方法.如王建会和孙国祥[20]取5粒双黄连胶囊的内容物,称质量后置于25 mL容量瓶中,甲醇溶解,超声分散30 min后冷却至室温,再用甲醇定容至刻度,离心后取上清液,用0.45 μm滤膜过滤得到待测溶液;张红伟等[21]取胶囊内容物1.0 g,用70%乙醇加热回流,过滤后将沉淀洗涤2次,合并上清液,通过AB-8型大孔径树脂柱分离纯化,用水和70%乙醇梯度洗脱,洗脱液浓缩成干膏,再用甲醇溶解定容至100 mL,最后以0.20 μm滤膜过滤得供试液.
1.6 凝胶剂
双黄连凝胶剂型样品需用适宜溶剂超声溶解,再经过滤后备用.如陈两绵等[22]取双黄连凝胶剂约0.7 g,置于50 mL容量瓶中,加适量50%甲醇,超声使其溶解,放置室温后,用50%甲醇定容至刻度,摇匀后0.45 μm微孔滤膜滤过,取滤液作为供试品溶液;赵永慧等[23]同样选取双黄连凝胶为样品,处理步骤与上述类似,不同的是溶剂使用乙腈/水(体积比为27∶73)进行溶解,得到待测样品.
2 色谱指纹图谱
色谱指纹图谱分析双黄连的方法主要有液相色谱(liquid chromatography,LC)及其联用、薄层色谱(thin-layer chromatography,TLC)、气相色谱(gas chromatography,GC)和毛细管电泳(capillary electrophoresis,CE)等方法.
2.1 LC
常用LC及联用方法主要包括高效液相色谱(high performance liquid chromatography,HPLC)、高效液相色谱-二极管阵列联用技术(HPLC-photo-diode array,HPLC-PDA)、高效液相色谱-质谱联用技术(HPLC-mass spectrometry,HPLC-MS)、高效液相色谱-电喷雾电离串联质谱(HPLC-electrospray ionization-MS,HPLC-ESI-MS)、超高效液相色谱(ultra performance liquid chromatography,UPLC)、超高效液相色谱-质谱联用技术(UPLC-MS)和超高效液相色谱-电喷雾电离串联质谱(UPLC-ESI-MS)等.
2.1.1 HPLC及其联用方法
HPLC因具有高压、高效和高灵敏度的优点而被广泛应用于中药质量控制检测[24-25].其原理是依据各组分在流动相中的吸附性能和分配系数不同而进行分离,并进入检测器检验,实现试样分析.至今,许多学者已采用该技术,对多种剂型的双黄连进行检测分析,为双黄连的质量控制带来了新的可能.以下围绕HPLC、HPLC-MS及HPLC-ESI-MS等方法在双黄连色谱指纹图谱分析中的研究情况展开综述.
陈俊[10]采用HPLC对10批双黄连口服液的药用成分进行了检验,在100.0 g双黄连产品中分析黄芩、连翘和金银花,3种主成分质量分数分别为25%、50%和25%,其中连翘的占比最大,且连翘苷和连翘脂苷在相应的范围内有良好的线性关系,研究得出不同批次的双黄连口服液品质差异较小;冯宏玲等[11]采用HPLC建立了双黄连口服液化学成分的指纹图谱,设置柱温为30℃,流动相为甲醇/冰乙酸,流速为1.0 mL/min,梯度洗脱,同时分离黄芩苷、连翘苷和绿原酸,其分析速率较药典法明显提高,以此提供了简便易行的双黄连多组分同时分离新方法;郭洁等[13]建立双黄连冻干粉指纹图谱并进行研究,以流动相乙腈/甲酸梯度洗脱,设置流速为1.0 mL/min、柱温为25℃、检测波长为280 nm、洗脱时间为25 min时,结果检测到21个色谱峰,并对7个色谱峰进行了归属,较为全面地反映了双黄连冻干粉所含的化学成分,有利于对其质量控制与评价;武煊[26]对10批兽用双黄连注射液HPLC指纹图谱进行了研究,流动相采用乙腈/乙酸,检测波长为280 nm,检测到16个共有峰,并对黄芩苷、连翘苷和绿原酸的含量进行了测定,该实验为后期中兽药的品质研究提供了很大的帮助;周志等[18]建立双黄连颗粒HPLC指纹图谱并进行探究(图1),流动相为乙腈/甲酸/水溶液,流速为1.0 mL/min,柱温为35℃,检测波长280 nm,检测出12批颗粒的图谱相似度≥0.998,得到共有色谱峰18个,确定了7个特征峰谱的归属,该方法稳定性好、重现性好,为双黄连颗粒质量控制与评价提供了依据.
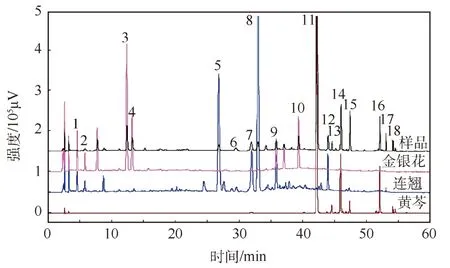
图1 双黄连颗粒与各单味药的高效液相色谱(HPLC)[18]
PDA目前已广泛应用于HPLC分析中,应用其作为检测器的优点是可进行全波长测定,并可在多通道模式进行分析,同时给出紫外吸收光谱和色谱图,便于组分定性和定量分析.王新宏等[12]对不同厂家的双黄连口服液和同一厂家的不同批号双黄连片剂的数字化色谱指纹图谱进行研究,以乙腈/乙酸/水溶液作流动相梯度洗脱,流速1.0 mL/min,柱温为室温,采用PDA检测器,在检测波长为280 nm时,鉴定了金银花、连翘和黄芩3种药材,且不同批号的图谱显示,每一批号片剂总色谱峰为28个,3个以上批号都出现的峰为23个,其色谱峰个数重叠率>82%,该实验采用1次进样进行测定,证实HPLC-PDA检测方法既简便又稳定,为双黄连制剂和中药材的测定提供可行的新方法.
HPLC-MS联用技术适用于极性强、挥发度低、相对分子质量大及热不稳定的混合有机物体系,更利于复杂物质的分离与检测.Han等[27]利用HPLCMS结合RBL-2H3/CMC柱层析,建立双黄连注射液中潜在过敏成分的二维分析体系,在保证具有良好重现性的基础上分析出双黄连潜在过敏成分——黄芩苷,该方法成功地实现了双黄连注射液中潜在过敏成分黄芩苷的筛选.
ESI是一种产生气相离子的软电离技术,可同时提供精确的相对分子质量和碎片结构信息,且可与各种色谱联用,特别适合于复杂样品分析.Zhang等[15]采用HPLC-ESI-MS建立双黄连注射液的色谱指纹图谱(图2),研究分析得出黄芩苷和芦丁都会促进组胺的释放进而引起过敏反应,此结果科学有效地揭示了双黄连注射液的过敏原因;罗奇志和罗佳波[28]利用HPLC-ESI-MS得到双黄连粉针剂的总离子流图,分析了双黄连粉针中的化学成分,采用全离子模式进行扫描,流动相为乙腈/水溶液,结果确定了18种化合物的相对分子质量,鉴定出9种化合物,除检测到已有报道的黄芩苷、连翘苷、绿原酸和咖啡酸等,还分析到未曾报道的奎尼酸(来自金银花);Luo等[29]利用HPLC-ESI-MS联用技术,在总离子流图中检测到43个色谱峰(图3),并对双黄连粉针剂中的各个成分进行了归属,其中21个峰来自金银花,20个峰来自连翘,4个峰来自黄芩(金银花和连翘有共有峰);王有志等[30]利用HPLC-ESI-MS首先得出奎宁酸标准品的MS图和离子扫描图(图4),依据标准曲线测得双黄连粉针剂供试液中奎宁酸的含量,为紫外吸收能力弱的奎尼酸的含量测定找到了可行的办法.
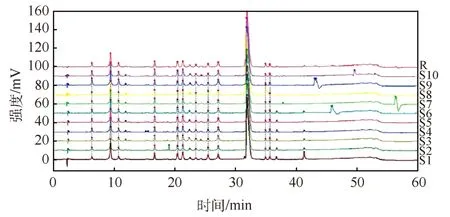
图2 不同批次双黄连注射液的HPLC指纹图谱[15]
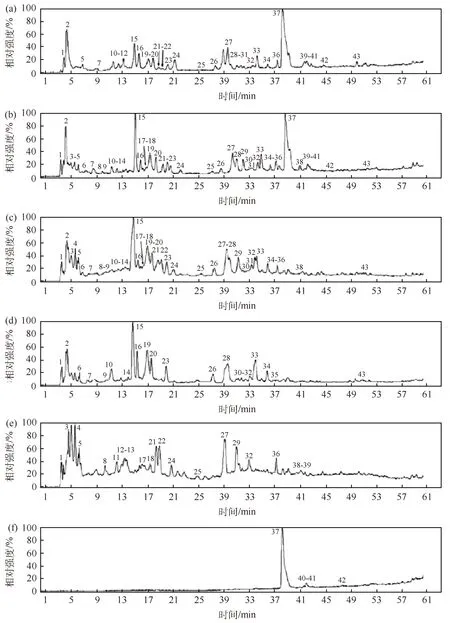
注:数字为峰编号.图3 不同性状双黄连的离子扫描[29](a)双黄连粉针供试液;(b)自制双黄连粉针提取物;(c)金银花与连翘合煎提取物;(d)金银花提取物;(e)连翘提取物;(f)黄芩提取物

图4 奎宁酸标准品的检测图谱[30]
2.1.2 UPLC及其联用方法
UPLC与HPLC的原理基本相同,但前者的分析速度更快,与MS联用后表现出更加优异的性能.石朗等[31]在对双黄连制剂进行UPLC-MS指纹图谱分析的同时,建立了双黄连中间体指纹图谱,在双黄连制剂和中间体图谱中都找到了指认的3个共有峰,表明其相关性良好,为双黄连高效、全面的质量控制提供了参考.
UPLC-MS同样可选用ESI作为电离源,提高分析的灵敏度和选择性.刘雪红等[32]建立双黄连口服液UPLC-MS指纹图谱,同时测定黄芩苷、连翘苷和绿原酸的含量,以甲酸/水/乙腈为流动相,在ESI负离子模式多反应监测模式下进行检测,相较之前HPLC或HPLC-MS技术,该方法可同时测定3种有效成分的含量,结果更快速、更高效、更准确且重现性好;张红伟等[21]同样采用 UPLC-ESI-MS方法,对以上3种有效成分同时测定,以醋酸钠/甲醇为流动相,在ESI正离子模式下进行检测,得出第1、2和3批双黄连胶囊提取液样品中绿原酸、黄芩苷和连翘苷的质量分数依次为 15.32、149.00和 4.05,16.05、158.00和 4.11,15.89、143.00 和4.14 mg/g,为双黄连胶囊的质量评价提供了简单、快速的检测方法;Yan等[33]利用UPLC-ESI-MS方法,在正离子和负离子2种ESI模式下对双黄连的成分进行检测,结果得到46个峰并对其进行了归属,结合背景减法为研究双黄连的代谢产物提供了依据,有助于检测双黄连的活性成分.
2.2 TLC
TLC是将适宜的固定相(如硅胶、氧化铝等)涂布在玻璃板或塑料等基片上,制成均匀薄层.待点样后并选用合适的展开剂进行展开后,与对照物的比移值进行对比分析的一种分离检测技术.该技术具有简便、高效的特点.
肖宏华等[34]利用TLC对双黄连口服液中的金银花进行鉴别,以乙酸乙酯/丙酮/甲酸/水(体积比为7∶3∶1∶1)作为展开剂,分别采用金银花中的木犀草苷和绿原酸,山银花中的灰毡毛忍冬皂苷乙、川续断皂苷乙作为特征成分,通过比较图谱中荧光条斑的有无和深浅,成功鉴别金银花中是否掺入山银花;程建国等[35]曾对《中华人民共和国兽药典2000年版》[36]和《中华人民共和国兽药典 2005年版》[37]收载的2种双黄连口服液成分的TLC方法进行了比较,结果前者的斑点更清晰,分离得更好,后者没有显示任何斑点,所以其推荐使用2000年版.李宇伟和连瑞丽[38]在用正交试验法优选双黄连口服液时,采用TLC对检测结果进行确证,分别对处方中的金银花、黄芩和连翘进行鉴别,TLC中供试品与对照品及对照药材色谱相应的位置上呈相同颜色的荧光斑点(图5),而阴性对照则无此斑点.该方法操作简便,重现性好,结果可靠,利于双黄连口服液及其中间产品的质量控制.

注:数字指不同样品点.图5 双黄连中3种中药材的TLC指纹图谱[38](a)金银花;(b)黄芩;(c)连翘
2.3 GC
GC法适用于有挥发性且不易分解的化合物的分离和分析,该方法具有分离效率高、速度快、样品量少、灵敏度高和选择性高等优点.
茹鑫等[39]建立GC指纹图谱和标准谱对照图(图6),对双黄连口服液中的可挥发物质进行检测和分析,结果共检测出96种挥发性物质,并归属了43种化合物,为双黄连的质量控制提供了有力依据;王栗等[40]利用GC法对双黄连粉针剂中的农药残留进行了测定,结果检测到连翘、黄芩和金银花中均含微量的有机氯农药,但远远低于我国对粮食中农药残留规定的标准,该方法利于双黄连的用药安全性检测.
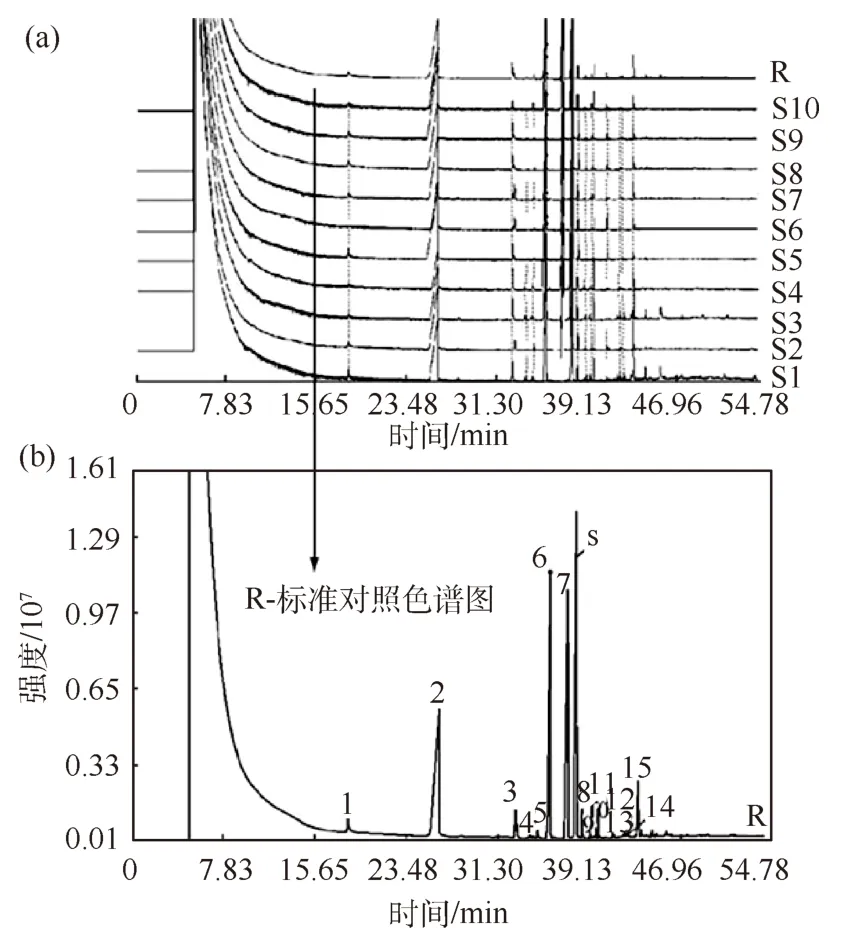
注:S1~S10是10批相同浓度的双黄连注射液编号;R是参比色谱.图6 双黄连注射液的检测图谱[39](a)气相色谱指纹图谱;(b)标准对照谱
2.4 CE
CE技术是依据样品中各组分之间的淌度和分配行为的差异来进行分离,主要用来分析在毛细管缓冲溶液中能离解为离子的物质.CE具有多种模式,如毛细管电泳色谱法(capillary electrochromatography,CEC)和胶束电动毛细管色谱法(micellar electrokinetic capillary chromatography,MEKC)等,该方法既能分离离子又能分离中性分子.
CEC是CE与HPLC相结合的一种快速、高效的分离方法,利用缓冲溶液的电渗流作为泵,具有操作简单、成本低和毛细管柱寿命长的优点,但是灵敏度稍逊于高效液相色谱.孙国祥等[41]建立了双黄连胶囊双波长CEC指纹图谱,以50 mmol/L硼砂(含20 mmol/L β环糊精和5%乙腈,溶液pH 7.55)为背景电解质,电压为12 kV,检测波长为203和256 nm,分别检测出16和11个共有指纹峰,为双黄连的质量控制提供了新依据;吕元琦等[42]建立CEC图(图7),分离并检测到以上3种成分,还测得了黄芩甙元的含量;Luan等[43]通过建立高效CEC方法,对双黄连中的重金属(Cd2+、Cr3+、Cu2+和 Zn2+)进行了有效测定,为双黄连中重金属离子的安全性进行把控.

图7 双黄连口服液毛细管电泳色谱[42]
MEKC是毛细管区带电泳与胶束液相色谱相结合而发展的方法,具有分离速度快、分离效率高和毛细管柱不易被污染等优点.Zhou等[44]采用MEKC对双黄连进行品质评价,以硼酸盐/二氢磷酸钠/十二烷基硫酸钠为缓冲液,甲醇和乙腈为有机改性剂,电压为15 kV.对双黄连中黄芩素、黄芩苷、绿原酸、汉黄芩素、黄芩素、连翘苷和金丝桃苷等7种主要成分进行定量测定,为提高双黄连制剂的整体分析和质量控制提供依据.
3 谱效学
谱效关系的研究是数学相关分析与药学的一个交叉应用,因此,一方面需要鉴定色谱指纹图谱中的主要化学成分,在此基础上通过谱效学研究揭示特征色谱峰;另一方面运用合理的数据处理分析方法,主要包括相关分析、回归分析、主成分分析、典型相关分析、聚类分析和灰度关联度分析等,实现数学相关分析与色谱指纹图谱的关联.目前,双黄连的谱效关系研究仍处于初步探索阶段,已形成一定的研究思路和模式.刘廷等[45]首次把HPLC色谱指纹图谱与双黄连对H1N1流感病毒致犬肾(Madin-Darby canine kidney,MDCK)细胞损伤的保护作用联系起来,通过HPLC数据和药效数据的SPSS分析,得出17个特征色谱共有峰,其中与该保护作用关系较大的峰有5个(3个峰对应金银花,2个峰对应连翘),得出双黄连中可以保护H1N1流感病毒致MDCK细胞损伤的成分为双黄连和连翘,该实验将双黄连的疗效与图谱结合,有效地分析出发挥药理作用的化学成分,为后期双黄连疗效与图谱的结合提供思路,并奠定了双黄连谱效学研究的基础;王建明和赵楚文[46]利用HPLC法测定双黄连中连翘苷的含量,再以大鼠为研究对象并设立实验组、模型组和对照组,采用不同方式处理后进行统计学分析,结果证实,先给予双黄连的大鼠在体温升高后有明显的下降趋势,验证了双黄连中的连翘苷成分有退热的效果,即该方法成功地探索出双黄连中与H1N1流感病毒致MDCK细胞损伤作用和退热作用相对应的有效成分.
双黄连作为一种中成药,其化学成分的构成并不能代表在体内产生生物效应的化学形式,色谱指纹图谱与谱效学的研究可以有效地阐释药物效应动力学,有利于双黄连品质的控制与评价.双黄连是一个多组分、多靶点和多作用途径的复杂体系,用精确的处理方法处理模糊的中药成分和药效关系尚且存在一定的局限性,色谱指纹图谱技术目前只能部分解决对共有峰成分的鉴定,体现某个组分与药效的相关性,而每个成分及成分之间的相互作用或协同作用没有阐述清楚,导致成分与功效只能达到部分相关,而未能实现完全相关.
2020年1月31日,中国科学院上海药物研究所和武汉病毒研究所开展的联合研究表明,双黄连口服液可抑制新型冠状病毒肺炎(COVID-19)[47],目前为止,该结果尚未纳入COVID-19的诊治方案中,其疗效有待于进一步的思考与研究.因此,进行双黄连的谱效学的研究是大有裨益的.目前,有关中药谱效学理论的研究方向及方法主要包括[48-51]:通过药物信息学方法寻找药效成分,建立多组分药效预测模型,对药效组分配伍进行优化设计,构建药效作用的多因素调节网络模型,探索多种药效组分协同作用机制;建立与动物(人)“证”模型相对应的状态函数关系式的现代中医药数理表述体系,根据特性蛋白质与效应体(药物)的齿合关系,按亲和色谱,以效应体靶向分离物——特性蛋白质为固定相,采用LC联用技术,建立品质或效应指纹图谱.因此,接下来的研究需要一方面建立双黄连多组分药效预测模型,对药效进行优化设计,探索多种药效组分协同作用机制;另一方面建立动物(人)“证”模型相对应的状态函数关系式,根据特性蛋白质与双黄连药效成分的齿合关系,采用LC联用技术建立质量或效应指纹图谱.
在现有的探索研究中,面临的难题主要包括:建立高重复性和高精度的适用于进行生物测定的方法难度高,这与生物个体的复杂性和待分析样品的复杂性有关;将指纹图谱所获取的化学成分特征信息与生物效应相关信息进行关联具有一定的困难;化合物对照品数量有限,部分样品价格昂贵;部分检验部门的硬件设施不够,难以满足实验要求.
4 结束语
不同剂型双黄连样品的前处理方法中,通常以甲醇/水进行溶解,也可选用乙腈/水体系,且多借助超声助溶,其中片剂和颗粒剂因其粒径相对较大,需研磨后再进行溶解,在色谱分析前多采用微孔滤膜进行过滤.色谱指纹图谱作为中药质量控制的关键技术在双黄连品质评价中有着举足轻重的地位,色谱相关的方法(LC、TLC、GC和CE)可以有效地对双黄连中间体及其成品进行成分鉴定和品质检测,可用于生产工艺与流程的监管.目前有关双黄连谱效关系的研究远不能满足实际需求.因此,仍需依据不同研究目的建立双黄连色谱指纹图谱与相应药效的关联,进一步探索其应用.
当前,中药色谱指纹图谱与谱效关系的研究逐渐增多,主要包括对中药的品质评价,提取与纯化,炒炭前后的作用等,但是由于谱效学的发展时间短暂,目前理论体系不够完善,技术尚有不足,从研究成分到药效再到临床应用之间尚有一定距离,未来仍需不断地发展与完善,为实现中药的现代化和国际化而努力.
