氨三乙酸强化零价铁/过一硫酸盐降解橙黄G
2021-04-30马红芳杨浩宇田委民伍凌斌陈秀峰
马红芳,杨浩宇,田委民,伍凌斌,陈秀峰,邹 景
氨三乙酸强化零价铁/过一硫酸盐降解橙黄G
马红芳1,2,杨浩宇1,田委民1,伍凌斌1,陈秀峰1,邹 景1*
(1.华侨大学土木工程学院市政工程系,福建 厦门 361021;2.华侨大学化工学院,福建 厦门 361021)
采用氨三乙酸(NTA)强化和改善零价铁/过一硫酸盐(Fe0/PMS)体系降解水中偶氮染料的氧化效能,以橙黄G(OG)为目标污染物,研究了NTA强化Fe0/PMS(NTA/Fe0/PMS)体系中OG的降解效果和NTA的强化作用机制,并考察了NTA、Fe0、PMS等主要反应物浓度和水中常见的共存物质对OG降解效能的影响.结果表明,NTA能够强化Fe0/PMS 体系降解OG的氧化效能,且初始pH值对其强化作用有显著影响.中性(pH=7)和酸性(pH=3)条件下, NTA/Fe0/PMS体系去除OG的表观速率常数分别较Fe0/PMS体系提高了31.3倍和5.5倍;增加NTA、Fe0和PMS浓度有助于OG的降解,但NTA超过8mmol/L或PMS超过1.0mmol/L时出现抑制现象;水质背景中, Cl-的存在促进了OG的降解, HCO3-、H2PO4-和腐殖酸则表现为不同程度的抑制作用;NTA/Fe0/PMS体系中,主导的活性物种为Fe0界面产生的SO4•-和•OH,界面作用和均相作用对OG的降解分别贡献了约83.2%和16.8%;加入NTA后,体系中生成的Fe3+/Fe2+能与其迅速形成络合物,既缓解了Fe0表面钝化层的形成,促进Fe0界面对PMS的直接活化,又提高了溶液中溶解性铁的浓度,促进均相作用对PMS的活化分解,使NTA/Fe0/PMS体系降解OG的氧化效能得到强化和改善.
氨三乙酸;零价铁;过一硫酸盐;活性物种;偶氮染料
偶氮染料被广泛用于纺织、造纸、皮革、化妆品和塑料工业等各个行业,大多偶氮染料具有毒性、致癌性和不可生物降解性,不能被传统的水处理技术有效去除,从而对生态系统和人类健康带来潜在的危害[1-2].因此,近年来许多学者积极探索高级氧化技术(AOPs)降解偶氮染料,如芬顿或类芬顿法[1-2]、臭氧法[3]以及光催化法[4]等.在诸多方法中,基于硫酸根自由基(SO4•-)的AOPs处理偶氮染料越来越受到人们的关注和重视[5-7].过一硫酸盐(HSO5−, PMS)是一种不对称的过氧化物,独特的结构使其成为生成SO4•-的常用氧化剂,而过渡金属Fe2+因活化能力较强、价格低廉、无毒安全等优点成为活化PMS广泛使用的活化剂[5-7].然而,Fe2+/PMS体系仍存在pH值适用范围过窄、氧化剂有效利用率低、体系持续氧化能力差等不足[6-7],从而使其应用受到限制.为此,研究者们使用零价铁(Fe0)作为Fe2+的来源活化PMS,有效缓解了Fe2+/PMS体系的不足[8-12]. Fe0活化PMS的反应主要包括Fe0表面腐蚀生成Fe2+[8-9]、Fe0通过电子转移诱导PMS分解生成SO4•-[12]、Fe2+均相活化PMS产生SO4•-、•OH和Fe3+[8-12]等过程.然而,随着反应的进行,Fe0表面会形成复杂的钝化膜[9],导致其活性点位和Fe2+的生成速度不断降低,从而造成Fe0活化PMS的效率和氧化效能下降[8].虽然采用超声、加热、紫外以及合成Fe0双金属材料[6-8,13]等方法可促进Fe0/PMS体系中Fe2+的生成,在一定程度上提高了Fe0/PMS体系对污染物的降解效果,但这些方法需要消耗额外的能量或存在有毒金属泄露造成污染的潜在风险,从而限制了其应用.
采用络合剂强化和改善基于Fe3+/Fe2+的芬顿或类芬顿均相体系的氧化效能已被广泛研究[1,14].加入的络合剂与体系中的Fe3+/Fe2+形成络合物,可增加Fe3+/Fe2+在中性pH值时的溶解度,拓宽体系的pH值应用范围;同时可减缓Fe2+与目标污染物竞争活性物种,提高体系的氧化效率[14].在Fe0/ PMS非均相体系中,加入络合剂理论上可以使生成的Fe3+避免水解及其后续反应,有可能减缓Fe0表面钝化膜的形成[9],强化和改善该体系的氧化效能,但目前还未见到这方面的研究报道.氨三乙酸(NTA)是一种络合能力很强的络合剂[15],且NTA及其与Fe3+/Fe2+形成的络合物易于生物降解[16].曾有学者研究了中性pH值条件下NTA对Fe0去除六价铬的影响,发现NTA的参与确实减缓了Fe0表面钝化膜的形成,提高了六价铬的去除效果[17].
基于此,本研究在Fe0/PMS体系中投加络合剂NTA,以典型的偶氮染料橙黄G作为模型污染物,系统探讨NTA对Fe0/PMS体系降解偶氮染料效能的影响.主要包括OG在NTA/Fe0/PMS体系中的降解效能, NTA强化Fe0/PMS的作用机制,以及反应物浓度和水质背景参数对OG降解效果的影响等,以期为NTA/Fe0/PMS的AOPs控制和消除水环境中的偶氮染料提供理论依据和技术参考.
1 材料与方法
1.1 材料与试剂
过一硫酸钾(PMS, KHSO5·0.5KHSO4·0.5K2SO4, KHSO5³47%)、零价铁粉(Fe0, 99.9%, 50nm)、叔丁醇((CH3)3COH)、甲醇(CH3OH)、苯酚(C6H5OH)、无水三氯化铁(FeCl3)、七水硫酸亚铁(Fe2SO4·7H2O)、1, 10-邻菲罗啉(C12H8N2.H2O )、乙酸钠(CH3COONa)、盐酸羟胺(NH3OHCl)、腐殖酸购买于阿拉丁试剂(上海)有限公司.橙黄G(OG, C16H10N2Na2O7S2)、氨三乙酸(NTA, N(CH2COOH)3)、氯化钠(NaCl)、磷酸二氢钠(NaH2PO4)、碳酸氢钠(NaHCO3)、氢氧化钠(NaOH)、高氯酸(HClO4)等购于上海国药集团化学试剂有限公司.所有试剂至少为分析纯,所有溶液采用超纯水配制.
1.2 实验过程
实验均在150mL的锥形瓶中进行,反应液体积为100mL.利用恒温振荡器(SHA-B)提供恒温水浴,控制反应温度为25℃,并设定搅拌强度为200r/ min.在锥形瓶中加入一定量的OG、NTA和PMS溶液及超纯水(自由基猝灭和水质背景影响实验,还需加入过量的清除剂(甲醇或叔丁醇或苯酚)或一定量的共存物质(Cl-、HCO3-、H2PO4-和腐殖酸)),并用NaOH和HClO4调节溶液pH值(整个过程不加缓冲溶液,也不再调整pH值),最后加入一定量的Fe0启动反应.在预设的时间点取样,立即用0.22μm的PES滤头过滤,并加过量的甲醇淬灭反应,然后测定OG、PMS和可溶性总铁浓度随着时间的变化情况.所有实验取3个平行样.
1.3 分析方法
OG浓度采用紫外可见分光光度计法测定,在478nm处测量试样滤液的吸光度值,通过吸光度与OG浓度的标准曲线求出.PMS浓度和溶液中可溶性总铁分别采用碘量法测定[18]和1,10邻菲罗啉法测定[19].
实验数据处理采用Origin8.5软件,曲线图和柱状图均采用Origin8.5软件做图.
2 结果与讨论
2.1 不同初始pH值条件下多种体系对OG的降解

[OG]0=0.2mmol/L, [PMS]0=2.0mmol/L, [NTA]0=1.0mmol/L, Fe00=0.112g (2.0mmol/L),=25℃
为了考察络合剂NTA对Fe0/PMS体系的影响,实验进行了多种体系在不同初始pH值条件下对OG的去除效果研究.如图1(a)所示,在pH=3的酸性条件下,反应进行到100min时,PMS、NTA以及NTA/PMS体系对OG几乎无降解,Fe0和NTA/Fe0体系对OG的去除率分别为22.4%和67.1%.与Fe0/ PMS体系中48.7%的OG去除率相比,NTA/Fe0/PMS体系的去除率提高到95%,说明NTA对Fe0/PMS去除OG的能力具有明显的强化作用.从图1(b)可知,在pH=7的中性条件下,Fe0、PMS、NTA一元体系和NTA/PMS、NTA/Fe0二元体系中的OG几乎不能去除.尽管Fe0/PMS体系对OG的降解仅有9.9%左右,但NTA/Fe0/PMS体系在180min内对OG的去除率达到了95.5%,说明NTA在中性条件下也可以强化Fe0/PMS体系的氧化能力.此外,由图1 (c)可知,pH=11.5的碱性条件下,多种体系对OG的降解情况与酸性和中性情况不完全相同.反应进行到60min时,虽然NTA、Fe0以及NTA/Fe0体系不能有效降解OG,但含有PMS的4个体系(PMS、NTA/PMS、Fe0/PMS和NTA/Fe0/PMS体系)对OG的去除则分别达到了93.6%、93.2%、88.6%和88.4%.研究报道,PMS在碱性条件下可以被快速活化产生单线态氧(1O2)和超氧自由基(O2•-)等活性物种,1O2和O2•-可去除含有不饱和官能团的染料等污染物[20].因此,在4个含有PMS的体系中,OG的降解应该源于碱激活PMS而产生的活性物种.本研究主要探查络合剂NTA对Fe0激活PMS降解污染物的影响,故只对酸性和中性条件下,Fe0/PMS和NTA/Fe0/PMS体系降解OG的动力学进行了拟合,拟合的假一级速率常数obs如图1(d)所示.可以看出,在pH=3和pH=7时,Fe0/PMS体系的obs值分别为6.7×10-3和0.63×10-3min-1,而NTA/Fe0/PMS体系的obs值则分别为37×10-3和19.7×10-3min-1,较Fe0/PMS体系分别提高了5.5倍和31.3倍.由此可见, NTA对Fe0/PMS体系的强化作用在中性环境条件下表现的更突出,同时也说明NTA在一定程度上拓展了Fe0/PMS体系应用的pH值范围.考虑到中性条件下去除污染物更具有实际应用意义,本文后续主要围绕pH=7的条件对OG降解性能进行研究.
2.2 NTA强化Fe0/PMS体系降解OG的机理
2.2.1 主要活性物种的识别 已有研究表明,Fe0/PMS体系中通常产生SO4•-和•OH等活性物种[6,8-12,21],进而以自由基机理氧化降解目标污染物.这些研究认为,Fe0表面腐蚀生成Fe2+(式(1)~(3)),并通过电子转移诱导PMS分解生成SO4•-(式(4));然后释放的Fe2+分解PMS生成SO4•-和•OH(式(5)~(6));生成的SO4•-与H2O、OH-作用转化为•OH(式(7~8)).然而,最新的研究表明,Fe2+激活PMS在酸性条件甚至是中性条件下生成的主要活性物种是高价铁(Fe(IV)),并不是长期以来被认为的SO4•-[22].Fe0/PMS体系中既然有Fe2+的释放,就有可能涉及到Fe(IV)的形成,而且Li等人[23]在Fe0/PMS氧化阿特拉津的研究中,也阐述了除SO4•-和•OH外, Fe(IV)也是体系中产生的主要活性物种.另外, Wang等人[24]在其最新研究中发现,虽然Fe2+激活过二硫酸盐(Fe2+/PDS)体系中产生的活性物种是Fe(IV)不是SO4•-,但投加络合剂后,络合剂与Fe2+之间的配位络合改变了活性物种的性质,且随着络合剂与Fe2+比值的提高,Fe2+/PDS体系中产生的活性物种逐渐由Fe(IV)转变为SO4•-(式(9))[25].本研究中,在NTA强化Fe0/PMS过程中既有Fe2+的释放,也会涉及NTA与Fe2+之间的络合,为了判断NTA/Fe0/PMS体系中的主要活性物种,实验进行了甲醇、叔丁醇和苯酚等对OG氧化降解的影响实验.
2Fe0+ O2+ 2H2O→ 2Fe2++ 4OH-(1)
Fe0+ 2H2O→ Fe2++2OH-+ H2(2)
Fe0+ HSO5-→ Fe2++ SO42-+ OH-(3)
Fe0+ 2HSO5-→ Fe2++ 2OH-+ 2SO4•-(4)
Fe2++ HSO5-→ Fe3++ SO4•-+ OH-(5)
Fe2++ HSO5-→ Fe3++•OH +SO42-(6)
H2O + SO4•-→ SO42-+•OH + H+(7)
OH-+ SO4•-→ SO42-+ •OH(8)

[OG]0=0.2mmol/L, [PMS]0=2.0mmol/L, [NTA]0=1.0mmol/L, Fe00=0.112g (2.0mmol/L), pH0=7,=25℃
Fe2+-L + S2O82-→ Fe3+-L + SO4•-+ SO42-(9)
甲醇与SO4•-和•OH之间反应的速率常数分别为1.1×107~7.7×107和9.7×108mol/(L×S)[11],二者均较高且较接近,所以甲醇通常被作为SO4•-和•OH很好的淬灭终止剂.而叔丁醇与•OH的反应速率常数为3.8×108~7.6×108mol/(L×S),远高于与SO4•-反应的速率常数4.0×105mol/(L×S)[10-11],因此叔丁醇常被作为•OH的有效淬灭剂.如在Fe0活化PMS降解双酚M的过程中,加入甲醇和叔丁醇使双酚M的降解分别受到85%和20%左右不同程度的抑制[11].然而,本研究中,甲醇和叔丁醇对NTA/Fe0/PMS体系降解OG的影响与以往文献报道的结果不一致[8,10-11,21].从图2(b)可看出,加入不同浓度的叔丁醇,即使叔丁醇与OG的物质的量比为500:1时,对OG的降解都无任何抑制作用,反而0.01mol/L叔丁醇表现出轻微的促进作用,促进作用可能与体系中生成的具有强还原能力的醇自由基(•CH2(CH3)2COH,0=-1.39~ -1.18V)对污染物的还原降解贡献有关[26].同时,由图2(a)还可知,1.0mol/L的甲醇对OG的降解仅有20%左右的抑制,远远低于以往文献的报道[11].上述现象出现的原因可能是体系中产生的SO4•-和•OH对OG的氧化降解主要发生在Fe0表面及近界面,或者是生成了非自由基Fe(IV)降解OG所致.因为甲醇和叔丁醇是亲水性物质,亲水性使其不容易扩散到Fe0表面,从而不能有效淬灭Fe0表面界面束缚的SO4•-和•OH[27].若体系中生成的主要活性物种是Fe(IV)而非自由基,Fe(IV)与甲醇和叔丁醇反应的速率常数较低,分别为5.72×102和6.0×101mol/(L×S)[23,28],因而也不能被甲醇和叔丁醇有效淬灭而消耗.苯酚是疏水性物质,可以快速扩散到固相Fe0表面,且与SO4•-和•OH等能快速反应,速率常数分别为6.6×109和8.8×109mol/(L×s)[29-30],但苯酚和Fe(IV)之间的反应速率常数仅仅为4.0×101mol/(L×s)[31].因此,苯酚对OG去除效果的影响可以用来区分体系中的活性物种主要为SO4•-和•OH还是Fe(IV).如图2(c)所示,苯酚的加入明显抑制了OG的降解,仅加入0.1mmol/L的苯酚(OG初始浓度的一半),OG的去除率降低至35.5%,加入0.5mmol/L的苯酚时OG的降解几乎完全被抑制.这说明了NTA/Fe0/PMS体系中OG的降解主要来自于SO4•-和•OH的自由基氧化途径,且反应主要发生在Fe0表面及其近界面.
2.2.2 NTA对Fe0/PMS体系的强化作用 为了探讨NTA对Fe0激活PMS的作用,试验考察了Fe0/PMS和NTA/Fe0/PMS体系中,溶解性铁和PMS浓度随着时间的变化情况,如图3所示.在Fe0/PMS体系中(图3(a)),溶解性铁的浓度在30min内逐渐增加到1.97μmol/L,后随反应有所微降,且整个反应过程中溶解性铁的浓度很低.而在NTA/Fe0/PMS体系中,溶解性铁的浓度随反应时间一直增加,180min时达到了13μmol/L,远高于Fe0/PMS体系.分析原因,主要是因为中性条件下,Fe0/PMS体系中Fe0的表面腐蚀过程比较微弱(式(1-4)),且随着反应进行,Fe0表面逐渐形成了复杂的钝化膜(式(10-12))[9],导致体系中Fe2+的生成速度不断降低.而NTA/Fe0/PMS体系中较高的溶解性铁主要源于以下方面:一是NTA有很强的络合能力,能迅速与体系中生成的Fe3+/Fe2+形成络合物,提高了溶解性铁的浓度[15,17];二是NTA的引入避免了Fe3+的水解及其与SO42-的进一步反应,使Fe0表面钝化层的形成得到缓解,从而促进Fe0释放生成Fe2+;三是钝化层的形成使Fe0具有一定程度的核壳结构,而NTA的加入促进了Fe0表面壳上的铁氧化物释放Fe2+/Fe3+.有研究报道,加入络合剂形成的络合物削弱了矿物表面的铁氧金属键,从而强化了金属离子的释放过程[32].此外,随着反应过程中PMS的分解, NTA/Fe0/PMS和Fe0/PMS体系的溶液pH值均呈现逐渐降低的趋势(反应结束时pH值分别为(3.25±0.02)和(4.80±0.02)).NTA/Fe0/PMS溶液的pH值因反应过程中PMS分解的更快(图3(b))而相对更低,这也有利于Fe0释放出更多的铁离子.Sun等[32]和Li等[17]在其研究中也报道了同样的现象,NTA的加入提高了Fe3O4/H2O2和Fe0/O2体系中溶解性铁的浓度.
Fe0+ SO42-+2H2O → FeSO4(s)+ 2OH-+H2(g)(10)
Fe3++ 2H2O→ FeOOH(s)+ 3H+(11)
FeOOH(s)+ SO42-+ H2O → FeSO4(s)+ 2OH-+
1/2H2(g) +1/2O2(g)(12)

[OG]0=0.2mmol/L, [PMS]0=2.0mmol/L, [NTA]0=1.0mmol/L, Fe00=0.112g (2.0mmol/L), pH0=7,=25℃
在NTA/Fe0/PMS体系中,随着Fe0的持续腐蚀和Fe2+的不断释放,PMS将被持续激活而分解.因此,与Fe0/PMS体系相比,NTA/Fe0/PMS体系中的PMS分解速率应该相对较高(图3(b)).可以看出,在考察的反应时间内,Fe0/PMS体系中仅有9%左右的PMS得到分解,而在NTA/Fe0/PMS体系中,约35%的PMS被活化分解.说明NTA的加入促进了Fe0/PMS体系中Fe0的不断腐蚀(式(1)-(4)),生成的Fe2+与NTA络合形成的NTA-Fe2+活化PMS生成SO4•-、•OH和NTA-Fe3+(式(13)-(14)),而NTA-Fe3+浓度的升高也能加快PMS的活化速率(式(15))[33],进而促进NTA/Fe0/PMS体系中PMS的分解.
NTA-Fe2++ HSO5-→ NTA-Fe3++ SO4•-+ OH-(13)
NTA-Fe2++ HSO5-→ NTA-Fe3++ •OH + SO42-(14)
NTA-Fe3++ HSO5-→ NTA-Fe2++ SO5•-+ SO42-(15)
综上,在NTA/Fe0/PMS体系中,NTA可以通过改善中性条件下Fe3+/Fe2+的稳定性、减缓Fe0表面钝化层的形成以及削弱Fe0表面上铁氧化物的铁氧键等作用促进Fe0不断腐蚀,既强化了Fe0对PMS的直接活化,又提高了溶液中溶解性铁的浓度,从而使更多的PMS分解生成更多的SO4•-和•OH等活性物种,进而强化了中性条件下Fe0/PMS体系降解OG的氧化效能.
2.2.3 NTA/Fe0/PMS体系中均相作用对OG降解的贡献 NTA的引入使Fe0/PMS体系产生了相对较多的可溶性铁(图3(a)),这些可溶性铁会均相活化溶液中的PMS产生自由基.为此,实验考察了NTA/Fe0/PMS体系中均相作用对OG降解的贡献.考虑到氧化体系中,Fe2+与NTA络合形成的NTA- Fe2+会被PMS迅速氧化生成NTA-Fe3+,故均相体系中的可溶性铁分别以Fe2+和Fe3+作为铁源,并根据反应结束时NTA/Fe0/PMS体系中可溶性铁的含量,将Fe2+和Fe3+浓度设定为0.013mmol/L,其他条件与NTA/Fe0/PMS体系相同.从图4可看出,NTA/ Fe2+/PMS和NTA/Fe3+/PMS体系对OG的去除情况较接近,反应180min时的去除率约为16.0%,远低于NTA/Fe0/PMS体系的95.5%.说明在NTA/Fe0/PMS体系中,OG的降解源于零价铁的界面作用和溶液中的均相作用,二者对OG的降解分别贡献了约83.2%和16.8%.可以看出,前者在体系中更占有优势,这与活性物种识别的结论相一致.另外,与Fe0/PMS体系9.90%的OG去除效果相比(图1(c)),均相体系相对较高的去除效果说明了NTA的引入同时强化了Fe0/PMS体系中的异相作用和均相作用.
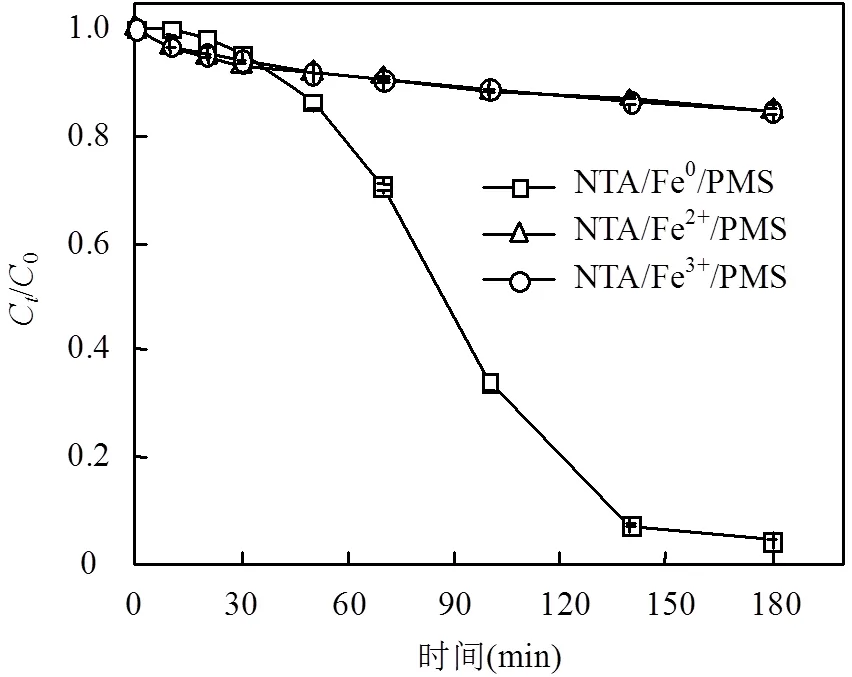
图4 均相作用对NTA/Fe0/PMS体系降解OG的贡献
OG]0=0.2mmol/L, [PMS]0=2.0mmol/L, [NTA]0=1.0mmol/L, Fe00=0.112g (2.0mmol/L),[Fe2+]0=0.013mmol/L, [Fe3+]0=0.013mmol/L, pH0=7,=25℃
2.2.4 NTA/Fe0/PMS体系中自由基的形成机理 基于上述实验结果和已有文献分析,在NTA/Fe0/ PMS体系中产生的主要活性物种为SO4•-和•OH,且体系中OG的降解主要发生在零价铁界面,SO4•-和•OH的可能形成机理如图5所示.

图5 NTA/Fe0/PMS体系中自由基的形成机理示意
一方面,在零价铁界面上,Fe0通过电子转移诱导PMS分解生成SO4•-和Fe2+(式(4)),并通过表面腐蚀生成Fe2+(式(1)-(3)).生成的Fe2+与零价铁界面上吸附的NTA形成界面络合物≡NTA-Fe2+,该络合物活化界面上吸附的PMS产生界面SO4•-和•OH(式(13)~(14)),部分SO4•-又与界面上的H2O和OH-结合生成了界面•OH(式(7-8)).同时,界面上新生成的≡NTA-Fe3+也会被Fe0还原为≡NTA-Fe2+,再继续活化PMS生成活性物种.尽管新生成的≡NTA-Fe3+也可活化界面吸附的PMS生成界面SO5•-(式(15)),但与式(13)-(14)相比反应速率很慢,而且SO5•-的氧化能力较低[23],故SO5•-不是零价铁界面上氧化降解OG的主要活性物种.
另一方面,零价铁界面上形成的部分≡NTA- Fe2+/≡NTA-Fe3+有可能释放进入均相溶液中,界面上的Fe2+也可能部分进入溶液与NTA络合,形成溶液中的NTA-Fe2+/NTA-Fe3+,进而激活溶液中的PMS产生SO4•-、•OH和SO5•-(式(13-15)),部分SO4•-与溶液中的H2O和OH-进一步反应生成•OH.同理,因反应速率和氧化能力较低,SO5•-不是溶液中降解OG的主要活性物种.
2.3 反应物浓度对NTA/Fe0/PMS体系降解OG的影响

图6 NTA浓度对NTA/Fe0/PMS体系降解OG的影响
[OG]0=0.2mmol/L, [PMS]0=2.0mmol/L, Fe00=0.112g(2.0mmol/L), pH0=7,=25℃
2.3.1 NTA浓度的影响 如图6所示,随着NTA浓度从0mmol/L增加到8mmol/L(NTA:Fe0物质的量比从0:1提高到4:1),NTA/Fe0/PMS体系的氧化效能明显提高,OG去除率达到95%以上的反应时间从130min缩短到70min,相应的obs值从0.63×10-3min-1增加到50.1×10-3min-1.然而,当NTA浓度进一步增加到10mmol/L(NTA:Fe0物质的量比为5:1)时,体系的氧化能力受到一定程度的抑制,obs值相应减小到38.3×10-3min-1.这种现象应该归因于过量的NTA淬灭消耗体系中的活性物种所致(如SO4•-与NTA反应,=5.89×107mol/(L×S))[34].与NTA有关的铁基类芬顿体系(如UVA/FeIII-NTA/S2O82-[34]、FeIII-NTA/ H2O2[14]等)降解污染物的研究也报道了相似的现象,但最佳的NTA:Fe物质的量比会随着铁的价态和剂量不同有所不同.如Sun等人[14]发现,随着Fe(Ⅲ)浓度从0.02mmol/L增加到0.18mmol/L时,最佳NTA:Fe(Ⅲ)物质的量比也相应地从1.5:1增加到4:1.
2.3.2 Fe0浓度的影响 如图7所示,当Fe0浓度从0mmol/L增加到4mmol/L(Fe0:PMS物质的量比从0:1增加到2:1),OG的去除效率分别为3.7%、93.8%、95.5%、99.8%,继续增加Fe0到10mmol/L (Fe0:PMS为5:1)时,OG完全降解的时间从180min逐渐缩短到50min.很明显,随着Fe0剂量的增加,OG的降解效果逐渐提高,obs值相应地从0mmol/L的0.3×10-3min-1提高到10mmol/L的69.3×10-3min-1.这是因为Fe0剂量的增加通常会释放较多的Fe2+而引起体系中NTA-Fe2+浓度的增加,较高的Fe0剂量和NTA-Fe2+浓度再去活化PMS使体系中产生了较多的SO4•-和•OH等活性物种(式(4),式(13~15)).然而,过量的Fe0也会引起电子浪费使电子利用效率降低[35],且产生过量的游离态Fe2+与SO4•-等发生淬灭反应[10],引起自由基的无效消耗.本研究中,当Fe0浓度超过4mmol/L(Fe0:PMS为2:1)时,OG降解效能随着Fe0浓度升高而提高的程度减弱.研究表明,在Fe2+/PMS均相体系中的最优Fe2+:PMS值为1:1[6],但在Fe0/PMS非均相体系中,为了获得满意的降解效果,优化后的最优Fe0:PMS值高达7.1:1[8]、11.9:1[21]、甚至44.6:1[9],远高于本文中的Fe0:PMS值.Barzegar等[8]也发现,US/Fe0/ PMS体系对污染物的降解效果随着Fe0浓度的提高而提高,当Fe0浓度超过一定值时,继续增加其浓度反而会降低污染物的降解效能.但在本研究考察的Fe0浓度范围(0~10mmol/L)内,未出现OG降解效能被过量Fe0抑制的现象.这可能与反应体系、反应条件以及污染物的种类不同有关.

图7 Fe0浓度对NTA/Fe0/PMS体系降解OG的影响
[OG]0=0.2mmol/L, [PMS]0=2.0mmol/L, [NTA]0=1mmol/L, pH0=7,=25℃
2.3.3 PMS浓度的影响 如图8所示,随着PMS浓度从0mmol/L增加到0.5mmol/L时,OG的降解效率从3.5%快速增加到93.7%,obs值相应从0.2×10-3min-1增加到25.3×10-3min-1.然而,当PMS浓度超过1.0mmol/L时,继续增加PMS浓度反而降低了OG的降解效果.在NTA/Fe0/PMS体系中,PMS是活性物种的产生源,当其他条件相同时,提高PMS浓度将会产生较多的活性物种从而提高OG的降解效能.然而,当PMS浓度超过一定值时,过量的PMS会与SO4•-等活性物种反应,产生活性比较小的活性物种SO5•-,从而限制了体系对OG的降解效果[21].此外,活性物种的自我消耗也是导致OG去除效果降低的原因[10].

图8 PMS浓度对NTA/Fe0/PMS体系降解OG的影响
[OG]0=0.2mmol/L, [NTA]0=1.0mmol/L, Fe00=0.112g(2.0mmol/L), pH0=7,=25℃
2.4 水体背景成分对NTA/Fe0/PMS体系降解OG的影响

[OG]0=0.2mmol/L, NTA0=1.0mmol/L, Fe00=0.112g(2.0mmol/L), [PMS]0=2.0mmol/L, pH0=7,=25℃
研究表明,水环境中的很多背景成分会干扰SO4•-等活性物种对有机物的降解[10].因此,本研究以水中常见无机阴离子(Cl-,HCO3-,H2PO4-)和天然大分子有机物腐殖酸(HA)作为代表,研究水体背景成分对NTA/Fe0/PMS体系中OG降解效果的影响,结果如图9所示.
2.4.1 氯离子(Cl-)的影响 从图9(a)可看出,Cl-为1mmol/L时对OG的降解基本无影响,提高其浓度至5mmol/L时产生一定的促进作用,继续提高至10mmol/L时其促进作用受到限制,说明Cl-对体系中OG的降解存在促进和抑制的双重作用.Cl-既可以促进零价铁的腐蚀[8]生成更多的活性物种,但也可与SO4•-等发生反应生成活性相对低的氯自由基(Cl•和Cl2•-).然而,研究报道[36],生成Cl•的正向反应与反向反应的速率常数大小约相当,因而Cl-和SO4•-之间的作用几乎不损失SO4•-的氧化能力.只有当Cl-以较高浓度(该浓度水平与体系中SO4•-的生成速率和条件有关[36])存在时,正向反应才占主导作用,产生对SO4•-更大程度的清除,从而表现出Cl-的抑制作用.本研究中,当在体系中加入1mmol/L的低浓度Cl-时,对零价铁腐蚀的促进影响极其轻微,因而OG的降解几乎无影响.随着Cl-浓度从1mmol/L升高到5mmol/L,其促进作用加强进而提高了OG的降解.当Cl-浓度继续提高至10mmol/L时,虽然可以进一步增强零价铁的腐蚀,但此时也出现了高浓度Cl-对SO4•-的淬灭而产生的抑制效应.
2.4.2 碳酸氢根(HCO3-)的影响 由图9(b)可知,HCO3-对OG的降解有较强的抑制作用.投加1mmol/L的HCO3-时,反应100min后OG的去除率从94%降低到25.7%.分析原因主要是HCO3-/CO32-的缓冲作用稳定了反应过程中的pH值,而pH值越低越有利于NTA/Fe0/PMS体系对OG的降解.在体系中分别加入1, 5和10mmol/L的HCO3-时,溶液pH值分别从最初的7.00左右降为反应结束时的5.70, 7.10和7.35左右,高于未投加HCO3-体系反应结束时的3.25.其次,体系中OG的降解主要发生在零价铁表面,而HCO3-容易吸附在零价铁表面占据其活化点位,并通过竞争吸附作用抑制零价铁表面OG的吸附,从而抑制OG的降解.此外,HCO3-是SO4•-的有效淬灭剂,与SO4•-反应(=(1.6±0.2)×106mol/(L×S))生成氧化能力较低的HCO3•(0=1.65V, pH=7),且HCO3•与OG的反应速率常数也较小[36],因而降低了对OG的氧化降解能力.HCO3-对氧化体系效能施加的负面影响在其他研究中也曾进行了报道[10,36].
2.4.3 磷酸氢根(H2PO4-)的影响 如图9(c)所示,加入1mmol/L的H2PO4-时,虽然对OG的降解产生了抑制作用,但反应180min后OG的去除率仍能达到93.6%,与未添加H2PO4-的去除效果无显著差别.继续增加H2PO4-浓度其抑制作用增强,加入5和10mmol/L H2PO4-的体系对OG的去除率分别降低到79.7%和32.2%.这种抑制作用在许多相关研究中也进行了讨论[8,10,37].一般认为,与HCO3-相类似, H2PO4-/HPO42-的存在对溶液pH值也有不同程度的缓冲作用[37].本文中,含有1, 5和10mmol/L H2PO4-的体系, 反应结束时的溶液pH值分别从最初的7.00左右变为3.85、6.58和7.35左右,而pH值越高越不利于体系中OG的降解.其次,H2PO4-与SO4•-反应生成氧化能力较低的H2PO4•(=7.0×106mol/ (L×S)).尽管H2PO4•也能与有机污染物发生反应,但其反应活性低于SO4•-,因而降低了整个体系的氧化能力[37].此外,H2PO4-易吸附在零价铁表面占据其活化点位,从而抑制了零价铁的催化活性[8,10].
2.4.4 腐殖酸(HA)的影响 由图9(d)可知,HA对OG的降解产生了轻微抑制作用,且随着HA浓度的增加,OG降解效率有所降低.当投加1~10mg/L的HA时,OG去除率均可以达到90%以上,继续提高至100mg/L时,去除率下降至81.6%左右.这种现象主要来自于两方面作用,一是HA在零价铁表面的竞争性吸附抑制了PMS的活化.Buffle等人[38]认为HA含有大量的官能团或吸附位,这种结构特性使其容易吸附在零价铁表面占据活化点位,使激活PMS生成活性物种的过程受到抑制.二是HA对SO4•-和•OH等活性物种具有一定的捕获能力(=(6.8±0.3)× 103L/(mgC·s),=(1.4±0.2)×104L/(mgC·s))[39],从而降低了OG的降解效果.有学者在其研究中也考察了HA对污染物降解效能的影响,结果表明HA表现出不同程度的负面影响[40].
3 结论
3.1 NTA能够强化Fe0/PMS体系降解OG的氧化效能,且强化效果受初始pH值的显著影响.在酸性(pH=3)和中性(pH=7)条件下,Fe0/PMS、NTA/Fe0/ PMS体系对OG的去除率分别为48.7%、95%和9.9%、95.5%,相应的obs分别为6.7×10-3, 37×10-3和0.63×10-3, 19.7×10-3min-1.加入NTA使Fe0/PMS体系的氧化效能分别提高了5.5倍和31.3倍,并拓宽了该体系应用的pH值范围.
3.2 NTA/Fe0/PMS体系降解OG的过程中,主要活性物种为Fe0界面上的SO4•-和•OH.加入NTA改善了中性条件下Fe3+的稳定性并缓解了Fe0的表面钝化,既强化了Fe0直接活化PMS,又促进其不断腐蚀生成Fe2+,显著提高了溶液中溶解性铁的浓度,从而促进PMS分解和活性物种的生成,使NTA/Fe0/PMS体系降解OG的氧化效能得到强化和改善.NTA/ Fe0/PMS体系中,Fe0的界面作用和溶液中的均相作用对OG的降解分别贡献了约83.2%和16.8%.
3.3 增加NTA投量及Fe0和PMS浓度有助于NTA/Fe0/PMS体系中OG的降解,但NTA超过8mmol/L或 PMS超过1.0mmol/L时对OG的降解产生了抑制.
3.4 水体背景成分对NTA/Fe0/PMS体系降解OG有不同程度的影响.其中,氯离子(Cl-,1~10mmol/L)起促进作用,碳酸氢根(HCO3-,1~10mmol/L)、磷酸氢根(H2PO4-,1~10mmol/L)和腐殖酸(HA,1~100mg/L)表现为不同程度的抑制.
[1] 毕 晨,施 周,周石庆,等. EGCG强化Fe2+/过硫酸盐体系降解金橙G的研究[J]. 中国环境科学, 2017,37(10):3722-3728.Bi Chen, Shi Zhou, Zhou Shiqing, et al. Degradation of orange G by Fe2+/peroxydisulfate system with enhance of EGCG[J]. China Environmental Science,2017,37(10):3722-3728.
[2] Cai M, Zhu Y, Wei Z, et al. Rapid decolorization of dye Orange G by microwave enhanced Fenton-like reaction with delafossite-type CuFeO2[J]. Science of the Total Environment, 2017,580:966-973.
[3] Bai C, Xiong X, Gong W, et al. Removal of rhodamine B by ozone-based advanced oxidation process [J]. Desalination. 2011,278 (1-3):84-90.
[4] Saleh T A, Gupta V K. Photo-catalyzed degradation of hazardous dye methyl orange by use of a composite catalyst consisting of multi- walled carbon nanotubes and titanium dioxide [J]. Journal of Colloid and Interface Science, 2012,371(1):101-106.
[5] Tan C, Dong Y, Shi L, et al. Degradation of Orange II in ferrous activated peroxymonosulfate system: Efficiency, situ EPR spin trapping and degradation pathway study [J]. Journal of the Taiwan Institute of Chemical Engineers, 2018,83:74-81.
[6] Ghanbari F, Moradi M. Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants: Review [J]. Chemical Engineering Journal, 2017,310:41- 62.
[7] Wang J, Wang S. Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants [J]. Chemical Engineering Journal, 2018,334:1502-1517.
[8] Barzegar G, Jorfi S, Zarezade V, et al. 4-Chlorophenol degradation using ultrasound/peroxymonosulfate/nanoscale zero valent iron: Reusability, identification of degradation intermediates and potential application for real wastewater [J]. Chemosphere, 2018,201:370-379.
[9] Tan C, Dong Y, Fu D, et al. Chloramphenicol removal by zero valent iron activated peroxymonosulfate system: Kinetics and mechanism of radical generation [J]. Chemical Engineering Journal, 2018,334:1006- 1015.
[10] Cao J, Lai L, Lai B, et al. Degradation of tetracycline by peroxymonosulfate activated with zero-valent iron: Performance, intermediates, toxicity and mechanism [J]. Chemical Engineering Journal, 2019,364:45-56.
[11] Yao J, Gao M, Guo X, et al. Enhanced degradation performance of bisphenol M using peroxymonosulfate activated by zero-valent iron in aqueous solution: Kinetic study and product identification [J]. Chemosphere, 2019,221:314-323.
[12] Yang Y, Guo H, Zhang Y, et al. Analysis on the removal of ammonia nitrogen using peroxymonosulfate activated by nanoparticulate zero-valent iron [J]. Chemical Papers, 2017,71(8):1497-1505.
[13] Li Z, Luo S, Yang Y, et al. Highly efficient degradation of trichloroethylene in groundwater based on peroxymonosulfate activation by bentonite supported Fe/Ni bimetallic nanoparticle [J]. Chemosphere, 2019,216:499-506.
[14] Sun S, Zeng X, Lemley A T. Kinetics and mechanism of carbamazepine degradation by a modified Fenton-like reaction with ferric-nitrilotriacetate complexes [J]. Journal of Hazardous Materials, 2013,252-253:155-165.
[15] Motekaitis R J, Martell A E. The iron(III) and iron(II) complexes of nitrilotriacetic acid [J]. Journal of Coordination Chemistry, 1994,31(1): 67-78.
[16] Nancharaiah Y V, Schwarzenbeck N, Mohan T V K, et al. Biodegradation of nitrilotriacetic acid (NTA) and ferric-NTA complex by aerobic microbial granules [J]. Water Research, 2006,40(8):1539- 1546.
[17] Li J, Chen Z, Shen J, et al. Influence of phosphate, citrate and nitrilotriacetic acid on the removal of aqueous hexavalent chromium by zero-valent iron at circumneutral pH [J]. Journal of the Taiwan Institute of Chemical Engineers, 2017,80:269-275.
[18] Liang C, Huang C, Mohanty N, et al. A rapid spectrophotometric determination of persulfate anion in ISCO [J]. Chemosphere, 2008,73(9):1540-1543.
[19] Harvey A E, Smart J A, Amis E S. Simultaneous Spectrophotometric Determination of Iron(II) and Total Iron with 1,10-Phenanthroline [J]. Analytical Chemistry, 1955,27(1):26-29.
[20] Qi C, Liu X, Ma J, et al. Activation of peroxymonosulfate by base: Implications for the degradation of organic pollutants [J]. Chemosphere, 2016,151:280-288.
[21] Kang Y, Yoon H, Lee W, et al. Comparative study of peroxide oxidants activated by nZVI: Removal of 1,4-Dioxane and arsenic(III) in contaminated waters [J]. Chemical Engineering Journal, 2018,334: 2511-2519.
[22] Wang Z, Qiu W, Pang S, et al. Further understanding the involvement of Fe(IV) in peroxydisulfate and peroxymonosulfate activation by Fe(II) for oxidative water treatment [J]. Chemical Engineering Journal, 2019,371:842-847.
[23] Li X, Liu X, Lin C, et al. Catalytic oxidation of contaminants by Fe0activated peroxymonosulfate process: Fe(IV) involvement, degradation intermediates and toxicity evaluation [J]. Chemical Engineering Journal, 2020,382:123013.
[24] Wang Z, Jiang J, Pang S, et al. Is Sulfate Radical Really Generated from Peroxydisulfate Activated by Iron(II) for Environmental Decontamination? [J]. Environmental Science & Technology, 2018, 52(19):11276-11284.
[25] Wang Z, Qiu W, Pang S, et al. Effect of chelators on the production and nature of the reactive intermediates formed in Fe(II) activated peroxydisulfate and hydrogen peroxide processes [J]. Water Research, 2019,164:114957.
[26] Fang G, Wu W, Liu C, et al. Activation of persulfate with vanadium species for PCBs degradation: A mechanistic study [J]. Applied Catalysis B: Environmental, 2017,202:1-11.
[27] Yang S, Xiao T, Zhang J, et al. Activated carbon fiber as heterogeneous catalyst of peroxymonosulfate activation for efficient degradation of Acid Orange 7in aqueous solution [J]. Separation and Purification Technology, 2015,143:19-26.
[28] Pestovsky O, Bakac A. Reactivity of Aqueous Fe(IV) in Hydride and Hydrogen Atom Transfer Reactions [J]. Journal of the American Chemical Society, 2004,126(42):13757-13764.
[29] Lindsey M E, Tarr M A. Inhibition of Hydroxyl Radical Reaction with Aromatics by Dissolved Natural Organic Matter [J]. Environmental Science & Technology, 2000,34(3):444-449.
[30] Ziajka J, Pasiuk-Bronikowska W. Rate constants for atmospheric trace organics scavenging SO4-in the Fe-catalysed autoxidation of S(IV) [J]. Atmospheric Environment, 2005,39(8):1431-1438.
[31] Chen J, Qi Y, Pan X, et al. Mechanistic insights into the reactivity of Ferrate(VI) with phenolic compounds and the formation of coupling products [J]. Water Research, 2019,158:338-349.
[32] Sun S, Zeng X, Li C, et al. Enhanced heterogeneous and homogeneous Fenton-like degradation of carbamazepine by nano-Fe3O4/H2O2with nitrilotriacetic acid [J]. Chemical Engineering Journal, 2014,244: 44-49.
[33] 潘 超.氨基三乙酸强化Fe(Ⅲ)/KHSO5去除4-氯酚的效能研究[D]. 哈尔滨:哈尔滨工业大学, 2013.Pan Chao. Nitrilotriacetate enhanced Fe3+/KHSO5oxidation of 4-chlorophenol in water [D]. Harbin: Harbin Institute of Technology, 2013.
[34] Jin Y, Wang X, Sun S, et al. Hydroxyl and sulfate radicals formation in UVA/FeIII-NTA/S2O82-system: Mechanism and effectiveness in carbamazepine degradation at initial neutral pH [J]. Chemical Engineering Journal, 2019,368:541-552.
[35] Liu H, Wang Q, Wang C, et al. Electron efficiency of zero-valent iron for groundwater remediation and wastewater treatment [J]. Chemical Engineering Journal, 2013,215-216:90-95.
[36] Liang C, Wang Z, Mohanty N. Influences of carbonate and chloride ions on persulfate oxidation of trichloroethylene at 20°C [J]. Science of the Total Environment, 2006,370(2/3):271-277.
[37] Wei X, Gao N, Li C, et al. Zero-valent iron (ZVI) activation of persulfate (PS) for oxidation of bentazon in water [J]. Chemical Engineering Journal, 2016,285:660-670.
[38] Buffle J, Greter F L, Haerdi W. Measurement of complexation properties of humic and fulvic acids in natural waters with lead and copper ion-selective electrodes [J]. Analytical chemistry, 1977,49(2): 216-222.
[39] Holger V L, Stephanie B, Insa R, et al. Degradation of chlorotriazine pesticides by sulfate radicals and the influence of organic matter [J]. Environmental Science & Technology, 2015,49(3):1673-1680.
[40] 孙绍芳,李佳龙,邱 琪,等. Fe(Ⅵ)/Na2SO3体系降解阿特拉津效能[J]. 中国环境科学, 2021,41(1):192-198.Sun Shaofang, Li Jialong, Qiu Qi, et al. Degradation efficiency of atrazine by Fe(VI)/Na2SO3system [J]. China Environmental Science, 2021,41(1):192-198.
Degradation of Orange G by Fe0/peroxymonosulfate with nitrilotriacetic acid enhancement.
MA Hong-fang1,2, YANG Hao-yu1, TIAN Wei-min1, WU Ling-bin1, CHEN Xiu-feng1, ZOU Jing1*
(1.Department of Municipal Engineering, College of Civil Engineering, Huaqiao University, Xiamen 361021, China;2.College of Chemical Engineering, Huaqiao University, Xiamen 361021, China)., 2021,41(4):1597~1607
Nitrilotriacetic acid (NTA) was applied to enhance and improve the oxidation efficiency of azo dyes in Fe0/PMS system. Taking Orange G (OG) as the target pollutant, the degradation efficiency of OG, the strengthening role of NTA, the effects of NTA, Fe0, PMS and general coexistence substances in water on the degradation of OG were studied in Fe0/PMS system enhanced with NTA(NTA/Fe0/PMS).The results demonstrated that the introduction of NTA could enhance the degradation of OG by Fe0/PMS system, and the initial pH had a significant effect on the enhancement. Under neutral (pH=7) and acidic (pH=3) conditions, the apparent rate constants of OG removal by NTA/Fe0/PMS system were 31.3 times and 5.5 times higher than those of Fe0/PMS system, respectively. Increasing the concentration of NTA, Fe0and PMS facilitated the degradation of OG, but negative effects were observed when NTA or PMS concentration were over 8mmol/L and 1.0mmol/L, respectively. In the context of water quality, the presence of Cl-promoted the degradation of OG, while HCO3-, H2PO4-and Humic Acid showed different degrees of inhibition. In NTA/Fe0/PMS system, SO4•-and •OH produced at Fe0interface was confirmed to be the dominant active species responsible for OG degradation, and heterogeneous and homogeneous activation of PMS contributed about 83.2% and 16.8% to the degradation of OG, respectively. When adding NTA to Fe0/PMS system, Fe3+/Fe2+generated could be complexed rapidly with NTA, which not only alleviated the formation of passive layer and promoting the direct activation of PMS on Fe0interface, but also increased the concentration of soluble iron in system, promoting the activation of PMS by homogeneous interaction. Thus, the degradation effect of OG in Fe0/PMS system was enhanced by NTA.
nitrilotriacetic acid;zero valent iron;peroxymonosulfate;active species;azo dyes
X703.5
A
1000-6923(2021)04-1597-11
马红芳(1969-),女,山西运城人,副教授,在读博士,主要从事高级氧化技术和水污染控制研究.发表论文30余篇.
2020-08-24
国家自然科学基金资助项目(51708231);福建省自然科学基金资助项目(2018J05084)
* 责任作者, 副教授, zoujing@hqu.edu.cn
