Patient-specific induced pIuripotent stem ceIIs as “disease-in-adish”modeIs for inherited cardiomyopathies and channeIopathies -15 years of research
2021-04-28MirunaMihaelaMicheuAnaMariaRosca
Miruna Mihaela Micheu, Ana-Maria Rosca
Miruna MihaeIa Micheu, Department of Cardiology, Clinical Emergency Hospital of Bucharest, Bucharest 014452, Romania
Ana-Maria Rosca, Cell and Tissue Engineering Laboratory, Institute of Cellular Biology and Pathology "Nicolae Simionescu", Bucharest 050568, Romania
Abstract Among inherited cardiac conditions, a special place is kept by cardiomyopathies (CMPs) and channelopathies (CNPs), which pose a substantial healthcare burden due to the complexity of the therapeutic management and cause early mortality.Like other inherited cardiac conditions, genetic CMPs and CNPs exhibit incomplete penetrance and variable expressivity even within carriers of the same pathogenic deoxyribonucleic acid variant, challenging our understanding of the underlying pathogenic mechanisms.Until recently, the lack of accurate physiological preclinical models hindered the investigation of fundamental cellular and molecular mechanisms.The advent of induced pluripotent stem cell (iPSC) technology, along with advances in gene editing, offered unprecedented opportunities to explore hereditary CMPs and CNPs.Hallmark features of iPSCs include the ability to differentiate into unlimited numbers of cells from any of the three germ layers, genetic identity with the subject from whom they were derived, and ease of gene editing, all of which were used to generate “disease-in-a-dish” models of monogenic cardiac conditions.Functionally, iPSC-derived cardiomyocytes that faithfully recapitulate the patient-specific phenotype, allowed the study of disease mechanisms in an individual-/allele-specific manner, as well as the customization of therapeutic regimen.This review provides a synopsis of the most important iPSC-based models of CMPs and CNPs and the potential use for modeling disease mechanisms, personalized therapy and deoxyribonucleic acid variant functional annotation.
Key Words: Induced pluripotent stem cells; Cardiomyopathy; Channelopathy; Genes; Mutation; Deoxyribonucleic acid variants
INTRODUCTION
Stem cell technology is one of the most dynamic areas in modern research, holding a great potential to alleviate or even cure various diseases.Particularly, induced pluripotent stem cells (iPSCs) are of a great interest given that they share the benefits of embryonic stem cells but lack their downsides.First, similar to embryonic stem cells, iPSCs are able to differentiate into tissues derived from all three germ layers, bothin vitroandin vivo.Secondly, iPSCs-derived cells will be immunologically identical to the host, making the use of immunosuppression unnecessary.Thirdly, there are no bioethical issues with the use of iPSCs.These unique features endorse them an excellent candidate for a wide array of applications such as cardiotoxicity screening, drug discovery, disease modeling, and cell therapy.
Ever since their first mention in 2006[1], we have witnessed a mounting body of data related to this rapidly growing field.Progress has been made in reprogramming and differentiation methods.Strategies for improving the maturity of iPSC-derived cardiomyocytes (iPSC-CMs) have been tested, and new applications to manage cardiac diseases have been tested.A recent Scientific Statement from the American Heart Association acknowledges disease modeling as possibly the most productive use of iPSCs[2].Several key characteristics endorse iPSCs as an ideal candidate for generating “disease-in-a-dish” models, particularly with regard to monogenic conditions.First of all, each iPSC line has a donor-specific genetic profile.Secondly, when collected, iPSCs are devoid of many of the epigenetic modifications caused by environmental and lifestyle factors, thus enabling the study of the genetic contribution to the disease.This aspect is of a particular importance in the case of Mendelian cardiac maladies, which are characterized by variable clinical expression and incomplete penetrance as a consequence of complex interactions between genetic backgrounds and environmental disease modifiers[3].Thirdly, iPSCs are quite malleable to genetic modification; accordingly, by using appropriate genome editing tools such as TALENs and CRISPRCas9, the deoxyribonucleic acid (DNA) sequence can be altered either by introducing causal DNA mutations into wild-type iPSC lines, or by repairing the causative factor to achieve phenotypic rescue in differentiated cells[2,4].
Inherited cardiac conditions (ICCs) include a variety of genetic disorders that primarily affect the heart.Among ICCs, a special place is kept by cardiomyopathies (CMPs) and arrhythmic diseases (i.e.channelopathies), which pose a substantial healthcare burden due to the complexity of therapeutic management and occurrence early mortality.Importantly, sudden cardiac death is frequently the first expression of the disease.Understanding the underlying genetic cause is the centerpiece of a timely diagnosis and targeted treatment[5].
CMPs are characterized by both structural and functional abnormalities of the ventricular myocardium that are not explained by flow-limiting coronary artery disease or abnormal loading conditions, each entity having particular characteristics at macroscopic and molecular level[6].Based on morphology, hereditary CMPs comprise the following types: hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), restrictive cardiomyopathy (RCM), arrhythmogenic cardiomyopathy (ACM), and left ventricular noncompaction (LVNC).
Inherited channelopathies (CNPs) are primary electrical disorders caused by mutations in genes encoding cardiac ion channels or associated proteins.As a result, malfunction of specific ion channels or of intracellular calcium handling occur, leading to electrical instability and predisposition to malignant arrhythmias in the absence of structural heart disease[7,8].The main cardiac channelopathies associated with increased risk of sudden cardiac death are long QT syndrome (LQTS), short QT syndrome (SQTS), Brugada syndrome (BrS), and catecholaminergic polymorphic ventricular tachycardia (CPVT).As comprehensive reviews of the genetics and clinical presentation of various ICCs have been written by our group[3,9]and other groups[10-12], we briefly point out the core genes associated with the CMPs and CNPs discussed in the present paper (see Tables 1 and 2)[12-19].It is to be noted that there is considerable genetic overlap among different CMPs and CNPs (Figure 1A and B, respectively).
MODELING DISEASE-SPECIFIC MECHANISMS
Due to the potential to differentiate into functional cardiomyocytes (CMs) that recapitulate patient-specific phenotypes, human iPSCs provide an excellentin vitroplatform to decipher the underlying disease-specific mechanisms and efficiently study inherited CMPs and CNPs in an individualized manner.
Inherited cardiomyopathies
HCM and DCM are the most frequently encountered genetic CMPs in daily clinical practice, therefore, unsurprisingly, they have been the most studied iPSC-CM-based models.In a recent report, Eschenhagenet al[19]comparatively analyzed 38 original papers that reported the characteristics of iPSC-CMs obtained from patients with HCM/DCM or generated from iPSC lines in which a HCM or DCM mutation had been genetically introduced[19].In summary, compared with their respective controls, the main features exhibited by HCM iPSC-CMs were the following: larger cell size, increased nuclear localization of nuclear factor of activated T cells (NFAT, a transcription factor) and increasedMYH7(orMYH7/MYH6ratio) expression[20-24].The most constant aberration identified in DCM lines was reduced peak force development[25], the molecular mechanisms of which are discussed, and explain the main clinical presentation of the disease.As for similarities between the two considered diseases, three anomalies were the most documented: sarcomere disarray, increasedNPPA/NPPBgene expression, and arrhythmic behavior[20,24,26,27].
Although not constantly associated with alterations in contractile force or kinetics, abnormal calcium handling appears to be a key pathological mechanism observed in iPSC-CM models of HCM[20,21,24,28].Valuable insights related to the molecular mechanisms of HCM pathogenesis have been provided by Seegeret al[29]in a model of patient-derived iPSC-CMs harboring a premature stop codon in theMYBPC3gene.When compared with the isogenic mutation-corrected iPSC lines, in addition to aberrant calcium signaling, patient-derived iPSC-CMs displayed molecular dysregulations without haploinsufficiency of the MYBPC3 protein.This observation could challenge the existing dogma of haploinsufficiency as the underlying mechanisms for HCM caused byMYBPC3premature termination codon mutations.The specific molecular signature included dysregulation of genes involved in calcium handling (ATP2A2, ATP2B2, andCASQ1), cardiac hypertrophy (GP130, JAK2, RRAS, MEK1, TWEAKR, andNPPB), stress response (HSPB1, HSPB6, HSPB7, IGF1, andIGF2), and structural organization of sarcomeres and mechanosensors (CSRP3andTCAP).
Disturbed calcium signaling has been shown to be a central pathological mechanism of diastolic dysfunction in familial HCM lines with variations in theMYH7, MYBPC3, andTNNT2genes[30].Using comprehensive functional imaging analysis (i.e.calcium imaging and traction force microscopy), Wuet al[30]revealed that diastolic Ca2+overload and increased myofilament Ca2+sensitivity contribute to diastolic dysfunction and demonstrated for the first time that patient-specific iPSC-CMs can recapitulate diastolic dysfunction characteristics at the single-cell level.Furthermore, calcium homeostasis was restored by Ca2+and late Na+blockers (verapamil, dilitiazem, ranolazine, and electlazine), which was reflected in diastolic function improvement in HCM iPSC-CMs.

Table 1 Main genes associated with inherited cardiomyopathies
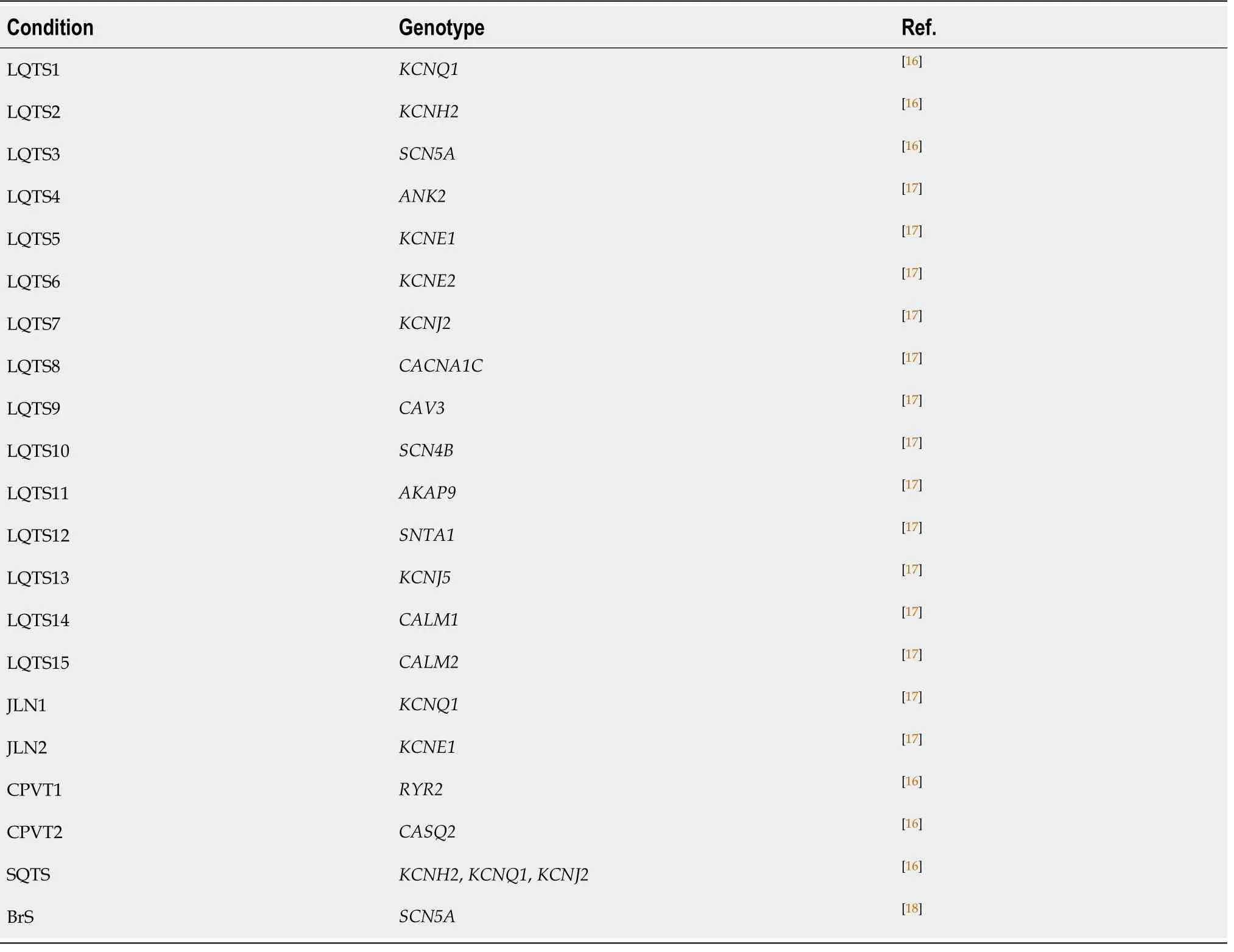
Table 2 Main genes associated with inherited channelopathies
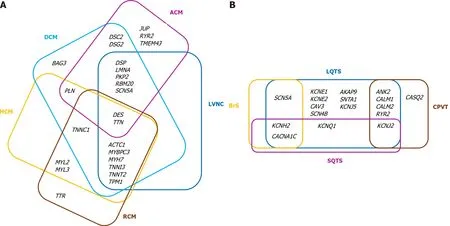
Figure 1 Diagram of the overlap of the main genes associated with inherited cardiac conditions.
More recently, it has been shown that molecular signaling differs within HCM iPSCCMs with diverse gene mutations.Isogenic models of HCM revealed differential phenotypes and mechanism-driven possible therapeutic targets inMYH7andACTC1cell lines, respectively[31].In spite of sharing key disease hallmarks, such as intracellular calcium overload and calcium transient arrhythmias, which were common in both models, modifications in contractility were entirely divergent, namely decreased contractility ofMYH7cells and gain of hypercontractility ofACTC1cells.Notably, the expression of Ca2+-binding proteins and hypertrophy-associated transcription factor activation also showed opposing behavior.Accordingly, compared with their respective controls, MYH7-R453C mutants were characterized by upregulation ofCASQ2, CALM1andCAMK2Dalong with MEF and NFAT nuclear translocation prompted byIRF8downregulation.Reversed expression patterns of those genes were found inACTC1-E99K iPSC-CMs.The study offered clinically-relevant data, given that the arrhythmogenic phenotype was rescued in both models following treatment with a mixture of dantrolene and ranolazine (a ryanodine receptor antagonist that inhibits sarcoplasmic Ca2+release into the cytosol and a late sodium current blocker promoting intracellular Ca2+efflux).In addition, the enhanced contractility displayed inACTC1iPSC-CMs was rescued by mavacamten (a selective allosteric inhibitor of cardiac myosin ATPase).Of note, an earlier study reported differences in the phenotypic features of iPSC-CMs obtained from a family with ACTC1-E99K mutation.These differences varied depending on the source subject, highlighting the value of isogenic iPSC-CMs in genotype-phenotype correlations.Thus, while arrhythmogenesis was manifested in allACTC1-E99K iPSC-CM lines, it was more common in cells obtained from the father, and less apparent in those derived from the two sons.This suggests that, in addition to the causative mutation, other factors (either genetic or epigenetic) might contribute to the disease phenotype[32].
The contractility consequences seem to be dependent on the specific DNA change rather than the affected gene.In contrast to the hypo-contractile phenotype described inMYH7-R453C mutants[31],MYH7-R403Q,MYH7-V606, andMYH7-R719 iPSC-CMs exhibited hyperdynamic contraction resulting from an increased proportion of myosin molecules in a disordered relaxed state conformation[33].Imbalance of myosin configurations (i.e.super and disordered relaxed states) led to the destabilization of interacting-heads motif interactions that were followed, in addition to increased contractility, by impaired relaxation, hypertrophic remodeling, excessive energy consumption, and metabolic stress.Mavacamten treatment restored the physiological super relaxed state/disordered relaxed state ratio and relieved downstream functional abnormalities, suggesting that chronic dysregulation of myosin conformations is a central mechanism of HCM.
Other reports have used iPSCs to decode genetic DCM physiopathology.The most frequently mutated gene in DCM isTTN, with truncating variants explaining up to 30% of familial cases[34,35].Other genes involved in DCM etiology are shown in Table 1.The most commonly encountered genes responsible for DCM areLMNA,DES,MYH7,MYH6,SCN5A,MYBPC3, andTNNT2, although at a lower prevalence thanTTN[36].Patient-derived iPSC-CMs have characteristics consistent with DCM, including sarcomere disorganization, reduced contractile function, and altered calcium handling[25,37-39].Hinsonet al[38]investigated the disease phenotypes of iPSCs engineered from DCM subjects with either truncating or missense mutations inTTN, and showing sarcomere insufficiency, impaired responses to mechanical and βadrenergic stress, and attenuated growth factor and cell signaling activation.
In a series of studies that focused onTNNT2-R173W missense mutation, iPSCCMs lines generated from affected family members exhibited a compromised ability to regulate calcium flux, reduced contraction force, and heterogeneous myofilament organization that were exacerbated by βadrenergic stimulation[25].In-depth analysis uncovered the epigenetic activation of phosphodiesterase genesPDE2AandPDE3Aas the underlying mechanism responsible for the defective βadrenergic signaling and contractile dysfunction[40].The latest data indicates thatTNNT2-R173W hampers molecular interactions between troponin and tropomyosin and restricts the binding of PKA to local sarcomere microdomains, resulting in diminished troponin phosphorylation and misalignment of sarcomeric proteins[41].An R173W variant also altered the interaction between sarcomere microdomain and cytoskeleton filamentsvia MYH7and AMPK, with consequent disturbance of sarcomere protein alignment and impaired contractility.In terms of phenotype rescue, AMPK activation by small molecules, such as A-769662, improved sarcomere-cytoskeleton attachment and partially recovered sarcomere protein misalignment and subsequent impaired contractility in mutated iPSC-CMs[41].
The phenotypes and the causal disease mechanisms linked toLMNAvariants have also been intensively studied.In iPSC-CMs with either a nonsense or missense mutation inLMNA, Siuet al[39]noticed enhanced nuclear senescence and augmented electrical stress-induced apoptosis that was significantly attenuated by pharmacological inhibition of the ERK1/2 pathway with the MEK1/2 inhibitors U0126 and selumetinib.Other investigators analyzed iPSC-CMs from three patients with distinctiveLMNAmutations (R225X, Q354, and T518fs)[42].Although all three types of diseased cells recapitulated the pathophysiological hallmarks ofLMNA-related DCM, the positive effect of ataluren treatment on the expression of full-length Lamin A/C, nuclear blebbing, apoptosis, and contractility was detected only in theLMNA-R225X mutant, suggesting that the effect might be codon selective[42].Other traits expressed byLMNA-mutated iPSC-CMs were bradyarrhythmia, beat rate variability, abnormal calcium handling, stress hypersensitivity, disorganized sarcomeres, and increased cell size[43,44].The incriminating mechanisms were aberrant activation of PDGF-signaling, which was rescued by pharmacological and molecular inhibition of PDGF receptor B[43], or epigenetic inhibition ofSCN5Athat was rescued bySCN5Aoverexpression[45].Mutations in otherDCM-related genes such asDES[37],MYH7[46],DMD[47,48],PLN[26], or even compound mutations have been modeled[49].
Characterization of iPSC-CMs from family members with LVNC who carried a nonsense variant in cardiac transcription factorTBX20, compared with iPSCCMs from unaffected relatives, revealed abnormal activation of transforming growth factor-β signaling leading to decreased cell proliferation capability.Moreover, inhibition of the transforming growth factor-β signaling pathway and correction ofTBX20alteration by CRISPR-Cas9 technology, successfully rectified the pathological phenotype[50].
Impairments in contractility, calcium handling, and metabolic activity have been nominated as key features in another model of iPSC-CMs generated from patients with delayed-onset LVNC caused by a missense mutation in theGATA4gene[51].
In an iPSC model with aTPM1-R178H mutation, it was shown that mislocalization of tropomyosin 1 was a central pathological change, triggering disturbance of the sarcomere structure and impaired calcium handling.Comprehensive analysis found involvement of complex molecular pathways centered on downregulation of the expression of numerous genes controlling heart development and positive regulation of cellular processes, including transcription factors (GATA4, GATA6) and sarcomeric proteins (MYBPC3, MYH6, TTN, TNNI3, TNNT2), thus linking sarcomeric dysfunction to LVNC[52].
ACM is another inherited CMP studied in iPSC models.In a series of reports published in 2013, CMs engineered from subjects having mutations in thePKP2gene efficiently recapitulated key disease features, including reduced cell surface localization of desmosomal proteins with altered desmosomal structure and a more adipogenic phenotype[53].These phenotypical changes were accompanied by upregulation of the pro-adipogenic transcription factor peroxisome proliferatoractivated receptor (PPAR)-γ and enhanced activation of respective signaling pathways[54,55].Furthermore, lipid droplets accumulation was prevented by administration of a specific inhibitor of glycogen synthase kinase 3β (6-bromoindirubin-3'-oxime)[55].Subsequent work revealed novel mechanistic insights in ACM pathogenesis or confirmed those already cited.Wenet al[56]reported that coactivation of normal PPAR-α and abnormal PPAR-γ pathways in ACM iPSC-CMs triggered markedly increased lipogenesis, apoptosis, Na+channel downregulation and defective intracellular calcium handling[56].In another iPSC-based model it was found that RhoA/ROCK signaling at the intercalated disc was essential for cardiomyocyte homeostasis[57].Using patient-derived iPSC-CMs with impaired cell-cell adhesion due to aPKP2frameshift mutation, or disturbed RhoA signaling caused by a nonsenseMYH10mutation, Dornet al[57]elegantly demonstrated that cardiomyocyte identity was lost following disruption of the RhoA/MRTF/SRF-signaling circuit.RhoA recruitment to cell-cell junctions was abridged in diseased cells, prompting increased levels of cytosolic G-actin and successive cytoplasmic sequestration of transcription factors such as MRTF that are involved in myocyte identity, preventing their entry into the nucleus.Finally, when exposed to an adipogenic environment, the mutated cells were poised to switch to a brown/beige adipocyte lineage, providing a possible molecular explanation of the clinical phenotype observed in ACM.Interestingly, a recent study reported lipid accumulation, increased pleomorphism, irregular Z-bands, and increased L-type calcium currents in iPSC-CMs carrying a novel frameshift mutation (L5218fs) in theOBSCNgene[58].The phenotypic alterations were accompanied by activation of adipocytokines and PPAR signaling pathways, diminished expression of the mutant protein and its anchor protein Ank1.5, in addition to downregulation of other desmosomal coding genes (PKP2, JUP, DSP)[58].
IPSC-CMs generated from an ACM patient with aDSG2mutation exhibited complex ion channel dysfunctions and abnormal cellular electrophysiology as well as increased sensitivity to adrenergic stimulation, indicating involvement of ion channel dysfunctions in arrhythmogenesis, independent of structural abnormalities[59].Subsequent work conducted by the same group established for the first time that enhancedNDPK-Bexpression,viaactivating SK4 channels, contributed to arrhythmogenesis inDSG2-related ACM, suggesting that NDPK-B could be a specific therapeutic target in selected patients[60].
Nget al[61]reported for the first time that some desmoplakin missense variants, such as DSP-R451G, are functionally equivalent to truncating alleles by promoting pathological vulnerability to calpain proteolysis and subsequent desmoplakin insufficiency.
There are few data related to restrictive cardiomyopathy modeling by iPSC.CMs harboring homozygousDES-Y122H mutation were reported to display abnormal desmin cytoplasmic aggregates responsible for the pathological phenotype[62].
Inherited channelopathies
A second category of genetic cardiac conditions extensively modeled using iPSC technology is represented by CNPs, for which electrophysiology studies exposed alterations of action potential, field potential, or Ca2+transients in engineered CMs.LQTS, in particular the first three types (LQTS1, LQTS2 and LQTS3), benefit from the most well-characterized iPSC-CMs models.Types LQTS that differ according to the underlying channel or gene mutation are shown in Table 1.
The first model of an arrhythmic syndrome using patient-specific iPSC-CMs was reported in 2010 by Morettiet al[63], who generated iPSCs from two affected members of a family with LQTS1 caused by a missense mutation (R190Q) in theKCNQ1gene.Mutant iPSC-CMs effectively reproduced the relevant features of the disease, namely prolongation of the action potential duration into atrial-like and ventricular-like cells, and increased occurrence of arrhythmic events when exposed to β-adrenergic agonists.Voltage clamp analysis revealed a substantial 70% to 80% reduction in the slowly activating delayed rectifier potassium currents (IKs) of LQTS1-iPSC-derived ventricular CMs due to a decreased number of functionalKCNQ1channels in the sarcolemma compared with the healthy counterpart.Beta-blocker treatment of LQTS1 CMs had a protective effect against catecholamine-induced arrhythmia.Similar findings have been reported in subsequent models of iPSC-CMs from LQTS1 withKCNQ1missense or frameshift mutations[64,65].Additionally, Wanget al[66]identified abnormalities in Ca2+handling linked to three distinctKCNQ1variants (R190Q, G269S, and G345E).CMs derived from all three edited iPSC lines displayed a characteristic LQTS phenotype and significant prolongation of the action potential duration compared with isogenic controls, which were corrected by treatment with L-type calcium channel antagonists.Similar results have been reported recently following an increase in the number of iPSC-CMs models of autosomal dominant, recessive, and compound heterozygous LQTS1[67-73], including analysis of models derived from specific populations[67,72].A plethora of LQTS2 iPSC-CMs models developed from patients harboring missense mutations inKCNH2have reproducibly shown prolonged action potential, increased arrhythmogenicity, and reduced rapidly activating delayed rectifier potassium current (IKr), compared with healthy control lines.The first analyzes of iPSC-based LQTS2 models were published in 2011 by two independent groups[74,75].Itzhakiet al[74]generated iPSC lines from a patient with aKCNH2-A614V variant.As expected, the derived-CMs exhibited substantial prolongation of the action potential, diminished IKr, early after depolarizations (EADs), and triggered arrhythmias.Specific pharmacological inhibition of IKr worsened the cellular phenotype, while administration of other pharmacological agents such as Ca2+channel blockers, or KATP-channel openers alleviated the pathological features[74].Thein vitromodel developed by the second group effectively replicated the variation in clinical phenotypes of two family members carrying the sameKCNH2-A561V mutation.Although the action potential duration was increased in the iPSC-CMs derived from both the clinically symptomatic patient and the clinically asymptomatic mother, an increased sensitivity (appearance of EADs) to stress (i.e.β-adrenoreceptor stimulation) was detected only in the symptomatic patient-derived cells[75].By comparing the electrophysiological properties of spontaneously beating CMs produced from LQTS2 cases and controls, it was suggested that cell-to-cell contacts in the syncytium result in compensatory mechanisms with a tendency to protect the repolarization system from major aberrations of physiological parameters.Although a considerable signal difference was detected between LQTS2 and control iPSC-CMs on single-cell patchclamp recordings (a 66% increase in action potential in LQTS2 cells), the differences were more modest (10%-20%) when using a microelectrode array technique on cell aggregates, similar to the surface electrocardiogram in respective patients[76].In later studies, the diseased phenotype was rescued either by genetic correction of theKCNH2mutation[77], or by allele-specific ribonucleic acid (RNA) interference, which selectively destroyed the mutant mRNA while leaving the wild-type mRNA undamaged[78].Various pharmacological agents were also shown to correct the electrophysiological anomalies[79-81], although for some molecules the effect was mutation-specific[82].An interesting observation was noted by Spenceret al[83], who established that in iPSC-CMs with aKCNH2-A422T mutation, the action potentials and intracellular calcium transients were prolonged in parallel.Furthermore, exposure to a Ca2+antagonist such as nifedipine, abbreviated the action potentials despite the IKr deficit.
Although abnormal calcium handling is common in both LQTS1 and LQTS2, there are major differences in this regard.Joutsijokiet al[84]used an innovative approach to differentiate the Ca2+transient statistics between these two LQTS-mutated iPSC-CMs.By combining machine learning and iPSC technology, the authors analyzed 90 LQTS1 transient signals from two cell lines and 138 LQTS2 signals from four cell lines, resulting in classification accuracies of up to 100%.The findings support the hypothesis that Ca2+transients are disease-specific or even mutation-specific.
Patient-specific iPSC-CMs models harboring gain-of-function mutations in theSCN5Agene efficaciously summarized LQTS3 pathognomonic electrophysiological traits, such as abnormal sodium currents and prolonged APD[85-88].A study by Malanat al[89]complemented prior findings by also showing a high incidence of EADs, a recognized trigger mechanism for arrhythmia, in disease cells.Treatment with mexiletine, specific sodium channel inhibitors, reduced action and field potential durations in LQT3 iPSC-CMs and alleviated EADs in a dose-dependent manner.Other types of LQTS, including LQTS7[90], LQTS8[91], LQTS14, and LQTS15[92-94]have been successfully investigated with iPSC technology.
CPVT comprises two main subtypes, CPTV1 (caused by mutations in theRYR2gene) and CPTV2 (determined by mutations in theCASQ2gene).Both genes are key regulators of CM calcium homeostasis, and dysfunction of either gene prompts abnormal intracellular Ca2+handling and signaling, and increased arrhythmogenicity under adrenergic stimulation.To date, numerous CPVT models have been developed using the iPSC platform, successfully recapitulating the arrhythmogenic phenotype seen in patients[95-104].In a study published in 2011, Fatimaet al[95]analyzed the functional properties of iPSC-CMs from healthy donors and a patient with CPVT1 carrying a novel heterozygous autosomal dominant mutation (RYR2-F2483I).Compared with healthy CMs, the mutated cells displayed arrhythmias and delayed afterdepolarizations (DADs), higher amplitudes and longer duration of spontaneous Ca2+release events in the basal state, as revealed by patch-clamp recordings and calcium imaging studies.Additionally, in CPVT-iPSC-CMs the Ca2+-induced Ca2+-release events continued after repolarization and were eliminated by increasing cytosolic cAMP levels with forskolin.In another CPVT1 model of iPSC-CMs harboring the RYR2-M4109R mutation, intracellular electrophysiological recordings evidenced increased development of DADs in CPVT-iPSCs-CMs compared with healthy CMs, which were further exacerbated by β-adrenergic stimulation; and, as opposed to previous findings, by forskolin.In contrast, thapsigargin (a specific inhibitor of the sarcoplasmic reticulum calcium ATPase pump) eradicated all afterdepolarizations in those cells, indicating that internal Ca2+stores play a central role in the pathogenesis of DADs.Indeed, laser-confocal Ca2+imaging revealed extensive whole-cell Ca2+anomalies (such as frequent local and large-storage Ca2+-release events, broad and double-humped transients, and triggered activity) that were aggravated under catecholaminergic stress and alleviated by beta-blockers.Also,RYR2-M4109R mutations significantly reduced the threshold for store-overload-induced Ca2+release[96].Dantrolene[97], flecainide[105], and S107 (an RYR stabilizer[104]) were other pharmacological agents shown to ameliorate the disease phenotypes.
More recently, the combined iPSC and CRISPR/Cas9 gene editing technics were used to validate preliminary data and, more importantly, to gain further insight into dysfunction produced by variations inRYR2gene.Weiet al[106], introduced a point mutation into wild-typeRYR2iPSCs by CRISPR/Cas9 gene editing.Similar to CMs generated from CPVT1 patient harboring F2483I-RyR2 mutation[101], edited iPSC-CMs carrying the same CPVT1-associated variant showed abnormal intracellular Ca2+handling, including longer and wandering Ca2+sparks, elevated diastolic Ca2+leaks, reduced sarcoplasmic reticulum (SR) Ca2+content, and increased susceptibility to arrhythmias caused by isoproterenol[106], suggesting that F2483I-RyR2 mutation produces leaky RyR2 channels.The same approach was used by Zhanget al[107]to assess aberrant Ca2+signaling and pharmacological sensitivity to three distinct CPVT1-associated mutations.While all three diseased iPSC-CM lines exhibited some abnormalities in calcium handling (i.e.irregular, long-lasting, spatially wandering Ca2+sparks and aberrant Ca2+releases), enhanced SR Ca2+leaks and diminished SR Ca2+contents were seen only in cells with Q4201R and F2483I, but not R420Q.Moreover, fractional Ca2+release and calcium-induced calcium release gain were higher in Q4201R than in R420Q and F2483I iPSC-CMs, emphasizing that Ca2+signaling abnormalities may vary depending on the mutation site.Several potential therapeutic interventions, including flecainide, dantrolene, and JTV519 (a Ca2+-dependent blocker of SERCA) were tested, indicating that drug sensitivities may also be mutation dependent.Using a wide-ranging methodology integrating optogenetics, tissue engineering, lab-on-a-chip technology, gene editing, and iPSC technology, Parket al[108]identified calmodulin-dependent protein kinase II activation as a key molecular pathway underlying exercise-triggered arrhythmia in CPVT patients, suggesting that its inhibition might be an effective therapeutic strategy in selected cases.
Recent data indicate thatRYR2screening should not be indicated only in subjects with stress- or exercise-induced symptoms.Using patient-specific iPSC-CMs, it has been shown thatRYR2-H29D variants elicit alteration of calcium homeostasis and molecular modifications such as aberrant SR Ca2+leak under physiological pacing, pro-arrhythmic electrical phenotypes, impaired and asynchronous contractile properties, and aberrant RyR2 post-translational modifications that occur only at rest[109].Furthermore, the authors hypothesized that the uncommon location ofRYR2-H29D mutations outside the four hot-spot regions linked to CPVT1, might be responsible for the distinct phenotypic expression.iPSC-based platforms have also been used to explore functional abnormalities in CMs generated from CPVT2 patients[99,102,110,111].Under beta-adrenergic stimulation, patient-derived iPSC-CMs carrying theCASQ2-D307H variant demonstrated disease-specific arrhythmogenic characteristics due to Ca2+-transient anomalies, afterdepolarization, reduced threshold for store overload-induced Ca2+-release, and upsurge of diastolic intracellular calcium concentration[99,102,110].
SQTS is a rare inheritable, autosomal dominant cardiac condition characterized by abnormally short QT intervals and an increased risk of atrial and ventricular tachyarrhythmias.The causal ion channel genes are shown in Table 2, variation inKCNH2being the most frequently observed in genotyped cases[7,112].The first SQTS model utilizing the iPSC platform was reported in 2018 by El-Battrawyet al[113].The authors generated iPSC-CMs from a patient with aKCNH2-N588K mutation and two healthy control subjects.Mutated cardiac myocytes exhibited enhanced IKr density and shortened APD compared with control cells, along with abnormal calcium transients and arhythmic propensity.Carbachol, an acetylcholine receptor agonist, increased the occurrence of arrhythmic events in diseased iPSC-CMs, while quinidine, and not sotalol or metoprolol, prolonged the APD and alleviated carbachol-prompted arrhythmias.In subsequent studies, patient-specific and gene-corrected iPSC-CMs were used to elucidate the SQTS phenotype either at single-cell[114]or tissue level[115].When compared with healthy control and gene-corrected CMs,KCNH2-T618I iPSCCMs were shown to display shortened APDs and increased beat-beat interval variability.Although no significant differences in totalKCNH2expression in control, gene-corrected, and SQTS iPSC-CMs were seen, membrane expression ofKCNH2was approximately 8-fold higher in mutated iPSC-CMs than in isogenic cells, suggesting that the aforesaid variant results in enhanced membrane expression ofKCNH2, which may contribute to the increased IKr density.Moreover, the phenotype was successfully rescued by BmKKx2, a short-peptide scorpion toxin acting as a selective IKr blocker[114].Shinnawiet al[115]examined conduction and arrhythmogenesis in confluent 2-dimensional iPSC-derived cardiac cell monolayers generated from a symptomatic SQTS patient also withKCNH2-N588K mutation.SQTS-iPSC-CM monolayers were characterized by abnormal repolarization properties and induced sustained re-entrant arrhythmias, while retaining a normal conduction appearance.
BrS is another cardiac channelopathy that has been modeled using iPSC technology.Various genes encoding either sodium, potassium, or calcium channels have been linked to BrS[116].Among them, theSCN5Agene was found to be most commonly mutated (Table 2).That gene encodes the alpha subunit of the main cardiac sodium channel (Nav1.5); loss of function variants result in reduced availability of functional Nav1.5 channels either through decreased plasma membrane channel expression or through altered channel gating properties[117].iPSC-CMs generated from BrS patients were shown to reflect the pro-arrhythmic phenotype associated with the disease and caused by blunted inward sodium currents, increased triggered activity, and calcium transient abnormalities.Daviset al[118]were the first to describe the molecular mechanisms that underlie BrS by using patient-specific iPSC-CMs harboringSCN5A_1795insD mutation, which effectively recapped the INa peak reduction and persistent INa associated with overlapped LQTS3/BrS[118].Another group investigated sodium currents, action potentials and calcium dynamics in iPSC-CMs derived from patients with type 1 BrS carrying two differentSCN5Avariants and in healthy control subjects[119].Mutated cardiac cells showed reductions in inward sodium current density, reduced maximal upstroke velocity of the action potential (AP), increased burden of triggered activity, abnormal calcium transients, and beating interval variation compared with control iPSC-CMs from healthy controls.Further analysis revealed markedly reduced expression ofKCNJ2, which encodes Kir2.1 inwardly rectifying potassium channel, an observation not previously described in BrS.Correction of the causative variant by CRISPR/Cas9-mediated genome editing prompted efficient resolution of triggered activity and abnormal Ca2+transients.
Additional data were provided by the work of Maet al[120], who reported that a repolarization deficit was involved in BrS.By comparing electrophysiological properties of iPSC-CMs generated from a patient carrying a compoundSCN5Amutation (A226V and R1629X) and a healthy sibling control, they observed an over 75% loss of sodium current in diseased cells.The decline in INa was reflected by altered action potential morphology with reduced maximum upstroke velocity and action potential amplitude at normal 1.0 Hz pacing frequency.At slow a slow pacing 0.1 Hz pacing frequency, an increased phase-1 repolarization action potential pattern characterized by a marked reduction of action potential duration and increased resting membrane potential occurred in a fraction of BrS CMs.Furthermore, disparities in the levels of transient outward K+currents (Ito) among the iPSC-CMs from either compound carriers or healthy controls were noticed, with 19% to 23% of the studied cells displaying high Ito densities.Importantly, in patient-derived iPSC-CMs, treatment with 4-Aminopyridine, an Ito blocker, completely reversed the increased phase-1 repolarization and restored the APD, indicating a coordinated role of INa and Ito in the arrhythmogenic mechanism of BrS.In-depth analysis of iPSC-CMs derived from two BrS subjects with anSCN5A-S1812X variant revealed reduced INa, amplified Ito, and increased ICaL window current probability along with conduction slowing, demonstrating coexistence of repolarization and depolarization impairment in diseased cells[121].
At present, it is widely acknowledged that patient-specific genetic background is a key determinant of the phenotypical manifestation of BrS, as was reported by a team of researchers from Spain and United Kingdom[122].As expected, iPSC-CMs from a patient with aSCN5Avariation recapitulated the loss of function of Nav1.5 associated with the syndrome.Also, a shift in both activation and inactivation voltagedependence curves and faster recovery from inactivation were reported.Remarkably, conventional heterologous expression systems (i.e.immortalized HEK293 cells coexpressing wild-type and mutant channels) failed to exhibit pro-arrhythmic changes in channel function, showing only a reduction in sodium current density, highlighting once again the need to assess the pathophysiological mechanisms of sodium channel mutations in a cardiac- and patient-specific model.
IPSC technology has also been used to model BrS caused by mutations in genes other thanSCN5A.Cerroneet al[123]were the first to describe the association of BrS and genetic variation inPKP2.They analyzed iPSC-CMs derived from five index cases carrying missense mutations inPKP2and perceived reduced sodium channel expression and current.The phenotype was rescued by transfection of wild-typePKP2,demonstrating that not only loss of PKP2, but also single amino acid mutations, can interfere with INa.
In the vast majority of clinically-diagnosed BrS cases (85%), the genetic cause is not known despite extensive use of NGS[124].Few research groups have used iPSC technology to uncover disease mechanisms at the cellular level in phenotype-positive genotype-negative patients[125,126].Notably, no clear cellular electrophysiological differences between the iPSC-CMs obtained from BrS patients without identified pathogenic mutations and control-derived cells were seen.That finding indicated that alternative pathophysiological mechanisms may be involved in those specific cases, such as right ventricular fibrosis or diminished cardiomyocyte coupling through gap junctions.Last but not least, BrS may be a multifactorial disorder, caused by an interaction of common genetic variations and environmental factors[125].
MODELING PATIENT-SPECIFIC THERAPEUTIC REGIMENS
The right dose of the right drug for the right patient at the right time is not only the mantra of personalized or precision medicine, but a common challenge faced daily by clinicians all over the world[2,127].With the advent of iPSC technology to guide therapeutic decisions in a patient-specific manner, tailoring treatment to a patient’s genetic background is yet to become a reality.
Prondzynskiet al[128]employed patient-specific iPSC-CMs to define disease-related mechanisms and also to guide treatment in an HCM-affected family carrying a novelACTN2missense mutation[128].Apart from previously described hallmarks of HCM, such as myofibrillar disarray, cell hypertrophy, increased myofilament Ca2+sensitivity, hypercontractility, and prolonged relaxation, iPSC-CMs demonstrated enhanced Ltype calcium channel current and prolonged action potential duration compared with isogenic controls.Following the beneficial results of improved contractile and electrophysiologicalin vitrophenotype with diltiazem, an L-type Ca2+channel blocker, the findings were translated into clinical settings where standard-dose diltiazem reverted the LQT phenotype in the son and sister of the index patient.
Although still in early stages, patient-derived iPSCs have been shown to facilitate optimal treatment in arrhythmic disorders.In a stepwise study, Terrenoireet al[88]established a patient-specific therapeutic regimen in a LQTS child with complex genetics and only partially-controlled arrhythmia with high-dose mexiletine[88].The index patient had ade novomutation in the sodium channelSCN5Aand a common polymorphism in the potassium channelKCNH2.First, electrophysiological analysis of the iPSC-CMs revealed that theSCN5Amutation was responsible for the patient’s symptoms.Furthermore, the authors found that mexiletine inhibited the IKr potassium channels in iPSC-CMs from both the father and the proband, irrespective ofKCNH2polymorphism, which explained the limited ability of mexiletine to completely correct the repolarization defect.Hence, alternative strategies to control INaL have been tested on patient-derived iPSC-CMs, such as changes in pacing rate or the addition of a second Na+channel blocker.The experimental data recommended mexiletine alone and an increased pacemaker rate as the best therapeutic option, which was further confirmed by the patient’s clinical evolution.In another LQTS3 model, mexiletine rescued the abnormal electrophysiology in iPSC-CMs from a patient harboring aSCN5Amutation (p.V1763M)[87].
Specific drug screening using patient-derived iPSC models has also been performed in CPVT, where β-blockers are the drugs of choice, but often fail to avoid malignant arrhythmias.In symptomatic CPVT patients under standard β-blocker treatment, it was shown that individual-specific iPSC-CMs had a subadequate antiarrhythmic response to β-blockers, while both patient and iPSC-CMs responded more effectively to flecainide[105,110].Clearly, the antiarrhythmic efficacy of different drugs is dependent on the underlying genetic variation.By patch-clamp analysis alone or by simultaneous patch-clamp and video imaging, Pölönenet al[129,130]assessed the antiarrhythmic effects of carvedilol and flecainide in CPVT patient-specific iPSC-CMs carrying diverseRYR2variants.They found mutation-specific differences in arrhythmias and drug responses, suggesting that proper treatment may vary even among subjects with mutations in the same genes[129,130].Evidence from earlier studies indicated that dantrolene was able to restore normal Ca2+spark properties and rescue the arrhythmogenic phenotype in a patient-specific iPSC model[97].Subsequent study revealed that not only that the location of the RYR2 mutation was critical for a favorable effect of dantrolene, but also suggested that the drug effect was dependent on the specific DNA alteration.Specifically, the antiarrhythmic effect was detected only in cases carrying mutations in the NH2-terminal or central regions of RYR2 protein.No effect was seen in subjects carrying mutations in the transmembrane region.Moreover, the effect of dantrolene was only minimal in iPSC-CMs with aQ4201Rvariant despite being located in the central region of RYR2 protein, even if at its end[131].Indeed, the dantrolene binding site is located in the NH2-terminal region ofRYR2between amino acid 601 and 620.After specific binding, the drug restores normal channel gating and prevents uncontrolled Ca2+release by stabilizing interdomain interactions between the NH2-terminal and central regions of RYR2, as previously reported[132,133].
MODELING VARIANTS OF UNCERTAIN SIGNIFICANCE
Sequencing of wide-ranging gene panels by high-throughput techniques on a daily basis has increased the rate of positive genetic testing, and it has also increased the detection of variants of uncertain significance (VUS).Recently, our group reported the yield of DNA testing in a cohort of HCM probands[134,135].Nearly half (45%) of the rare variants identified in our study were novel, and thus classified as of VUS.All but two were found only once in our cohort.Similar results were obtained in other studies, which reported a prevalence of 35%-40% of new mutations, half of which were unique for a family[136].The conclusive classification of VUS is encumbered by challenges, particularly in cases of “private” mutations, as it involves computational and population-based studies, not rarely misleading[137,138].Combined use of recent technologies such as iPSC and gene editing have enabled functional annotation in specific cases.
Lvet al[139]used a dual-integrase cassette exchange platform to rapidly and efficiently generate iPSCs with the TNNT2-E251D variant harbored by a woman with severe HCM and otherwise negative genetic testing.Although the mutation was generally predicted to be pathogenic byin silicoanalysis, the allele frequency of 0.03% in the Exome Aggregation Consortium database was inconsistent with the disease incidence (i.e.too high), and the ClinVar archive included conflicting interpretations of clinical significance, but mostly VUS.TNNT2-E251D iPSC-CMs had normal responses to isoproterenol, suggesting that the variant might not be pathogenic.To exclude the possibility that the failure in attaining a pathological phenotype was due to lack of a permissive genetic background in the studied cells, the authors introduced an E251D point mutation into an edited iPSC line known to be vulnerable to cardiomyopathy with CRISPR-Cas9.Comprehensive investigation of the E251D iPSC-CMs showed normal responses to isoproterenol and no significant increase in cell size or expression of genes previously reported to be upregulated in HCM iPSC-CMs (e.g.,TNNT2, MYL2, MYL4,andMYH7).This approach allowed specific recommendations to be made to relatives, namely not to undergo cascade genetic screening for theE251Dvariant.
In another study, iPSC-CMs were produced from an asymptomatic subject with a HCM associated mutation inMYL3,and reported by the ClinVar database to be likely pathogenic[23].Extensive assays, including measurement of gene expression, sarcomere structure, cell size, contractility, action potentials, and calcium handling, were performed on isogenic iPSC-CMs that were either corrected or carrying homozygous alleles found that the VUS was benign.
With the goal of functional prediction of pathogenicity, Pettinatoet al[140]developed a scalable human cardiomyocyte platform to interrogateTNNT2variants previously identified in the human population.Using iPSC-CMs in cardiac microtissue and single-cell assays, they examined 51TNNT2variants, including 30 pathogenic/likely pathogenic variants associated with HCM/DCM, and 21 VUS.Experimental evidence including transcriptomic changes and cardiac microtissue contraction, supported the reclassification of two pathogenic/likely pathogenic variants and two VUSs.These findings are of a great interest given that mostTNNT2variants identified in the human population are classified as of VUS.therefore definite reclassification would enable specific clinical decision making for individuals harboring these variants.
In a similar manner, iPSC models were used to decipher the pathogenicity of variants detected in patients with inherited CNPs.By combining patient-specific iPSCs and genome editing, Garget al[141]demonstrated the pathogenicity of a novel VUS in theKCNH2gene.Compared with healthy control cells, VUS iPSC-CMs displayed electrophysiological abnormalities consistent with LQTS2 phenotype (prolongation of action potential duration and reduced IKr density), which were rescued by VUS correction by CRISPR/Cas9.Furthermore, the introduction of the homozygousKCNH2-T983I variant in a healthy control line recapitulated the hallmark LQTS phenotype, confirming that the mutation was sufficient to prompt the disease.
Generation of iPSC lines from every single individual with a VUS in a CMP-/CNPrelated gene, followed by allele correction, and functional assessment is laborious and virtually impossible.Hence alternative approaches exploiting already existing and functionally characterized human iPSC lines has been considered.For example, commercially available human iPSC-CMs were used to screen aKCNJ2VUS detected in a LQTS7 proband by whole exome sequencing[142].VUS overexpression was associated with a substantial prolongation of APD with evidence of arrhythmic activity, emulating the clinical phenotype, and thus supporting causality of the variant.
Chavaliet al[143]established a patient-independent human iPSC model as a new tool for rapid determination of genetic variant pathogenicity in LQTS.The authors used CRISPR/Cas9 to introduce aCACNA1CVUS from an unrelated healthy volunteer into a previously established iPSC line.Functional changes detected in gene-edited iPSCCMs allowed reclassification ofCACNA1C-N639T variant to ‘’likely pathogenic”.
Considering all the available evidence, it can be easily seen that a screening platform based on edited human iPSC lines might be more informative than currently used procedures for variant classification, such as computational and populationbased methods.
CONCLUSION
Due to various genetic and environmental modulatory factors, Mendelian CMPs and CNPs are characterized by variable expressivity and incomplete penetrance, which often delays the clinical management of such patients.One issue to be addressed by upcoming studies is whether iPSCs can be used to identify genetic modifiers and to unveil the protective or aggravating underlying regulatory mechanisms.As a proof of concept, Chaiet al[144]used complementary physiological and genomic analyses to identify genetic modifiers explaining the variable expressivity observed in a large LQTS2 family.
Although the feasibility of this new technology for disease modeling and drug testing has been demonstrated, there are currently some limitations that should be addressed in order to further recommend the use of iPSC-CMs in clinical practice.Thus, the main setbacks in using this approach on a large scale are the reproducibility of results among multiple laboratories and the immature phenotype displayed by these cells.
The first is due to the use of various methods for inducing pluripotency, chromosomal instability throughout the reprogramming process andin vitromanipulation, the purity in myocyte composition, and batch disparities in differentiated CMs[145].Therefore, implementation of standardized protocols for patient-specific lines is important.Second, most iPSC-CMs have an immature structural and functional phenotype, with fetal gene expression, disorganized sarcomeres, primarily relying on glycolysis, and having contractile features different from those of adult CMs, such as spontaneous beating[146].Those properties could negatively impact the interpretation of the cellular responses to various drugs and the prediction of the clinical value of the respective compounds.Consequently, it is imperative to develop methods to generate CMs with a more mature phenotype in order to improve the predictive value ofin vitrostudies.Recently, important progress in the maturation of iPSC-CMs has been made by using small molecules[147], environmental manipulation[148]and three-dimensional culture[149].IPSC-based research is still at an early stage.Nevertheless, one can undoubtedly see its boundless potential for advancing personalized clinical management of individuals with inherited CMPs and CNPs.
杂志排行

World Journal of Stem Cells的其它文章
- Hypoxia-inducibIe factor-1α-mediated upreguIation of CD99 promotes the proIiferation of pIacentaI mesenchymaI stem ceIIs by reguIating ERK1/2
- Orthobiologics in the treatment of hip disorders
- Calcium channels and their role in regenerative medicine
- Stem cell therapy for heart failure: Medical breakthrough, or dead end?