Transformation of coccolithophorid Emiliania huxleyi harboring a marine virus ( Coccolithoviruses) serine palmitoyltransferase (SPT) gene by electroporation*
2021-04-14WeicongCAIXuetingWANGJinjingSUJianLIJunZENGGuilingLIJingwenLIU
Weicong CAI , Xueting WANG, Jinjing SU , Jian LI, Jun ZENG, Guiling LI,**,Jingwen LIU ,**
1 College of Food and Bioengineering, Jimei University, Xiamen 361021, China
2 Fujian Provincial Key Laboratory of Food Microbiology and Enzyme Engineering, Xiamen 361021, China
Abstract Emiliania huxleyi is the most prominent modern coccolithophore, a group of marine unicellular eukaryotes that play a critical role in ocean biogeochemistry. Coccolithoviruses are large double stranded DNA viruses, which is responsible for the demise oflarge oceanic blooms formed by E. huxleyi. E. huxleyi virus (EhVs) acquired a series of enzyme-coding genes predicted to be involved in the sphingolipid biosynthesis by horizontal gene transfer between virus-host. Currently, there is limited experimental validation identifying the functions of these genes in EhV. Genetic transformation of eukaryotic cells is a powerful tool to get an insight into gene functions of the studied organisms. Serine palmitoyltransferase (SPT) catalyzes the first committed step in de novo sphingolipid biosynthetic pathway. Here, a novel vector system for the transformation of E. huxleyi was designed. It contained fragments of promoter and terminator sequences of E. huxleyi endogenic fucoxanthin chlorophyll a/ c-binding protein gene “ fcp” and harbored EhV-99B1 spt gene. The resultant recombinant transformation vectors pEhux-I- spt and pEhux-II were co-transferred into E. huxleyi BOF92 by electroporation. Transformants were obtained upon glufosinate-ammonium selection,and confirmed by Southern hybridization, genome PCR, qRT-PCR and Western blot screening of spt gene,which indicated that spt gene was integrated into the nuclear genome and was expressed at the mRNA and protein levels. The expression of the viral spt gene led to differences in lipid compositions analyzed using thin-layer chromatography (TLC). The results present the genetic transformation system for E. huxleyi,providing additional genetic resource with potential for exploring basic biological questions such as the virus-host interactions.
Keyword: E miliania huxleyi; coccolithovirus; genetic transformation; serine palmitoyltransferase (SPT);total lipid
1 INTRODUCTION
Coccolithophore (haptophytes) is a group of globally important unicellular marine microalgae.One prominent feature of this alga is the ability to produce “the coccoliths”. It is considered the most productive calcifying organism on earth and becoming a major factor in the global carbonate cycle and climate changes (Westbroek et al., 1993; Read et al.,2013; Hernández et al., 2018). Emiliania huxleyi is one of the most abundant and widely distributed coccolithophore in modern oceans. It is considered the world’s major producer of calcite and an important factor in determining the exchange of CO2between the oceans and the sediments (Dymond and Lyle,1985), Since its ability to synthesis long-chain alkenones, as a suite of organic biomarkers providing a highly characteristic record in the sedimentary archive (Westbroek et al., 1993).
Viruses that infect phytoplankton play a key role in shaping the evolution and dynamics of the oceanic microscale ecosystem (Suttle, 2005). In natural marine ecosystem, some E. huxleyi species could be infected by the specific large dsDNA lytic viruses(EhVs, genus Coccolithovirus), and its bloom collapsing has been frequently linked to virus control in the marine environment (Bratbak et al., 1993;Brussaard, 2004). The EhV-86 virus genome sequence identified a series of enzyme-coding genes apparently involved in sphingolipid metabolism (Wilson et al.,2005). Phylogenetic evidence demonstrated the occurrence of horizontal gene transfert of these genes between E. huxleyi-EhV system (Monier et al., 2009).An unusual glucosylceramide was isolated from EhV-86 infected host cells (Rose et al., 2014), which attributed to coordinated interactons between hostand viral-encoded sphingolipid biosynthetic enzymes(Michaelson et al., 2010; Rosenwasser et al., 2014),providing a fascinating new paradigm for host-virus interactions. Sphingolipids are essential structural components of all eukaryotic membranes and are important signaling lipids in diverse cellular pathways,such as apoptosis, and they play a crucial role in the life cycle ofintracellular pathogens (Schneider-Schaulies and Schneider-Schaulies, 2015). It was attractive that the EhV-encoded serine palmitoyl transferase (vSPT), the first-rate limiting enzyme in de novo sphingolipid synthesis exhibited a novel metabolic strategy leading to the production of a unique suite of viral-specific glycosphingolipids(vGSLs) (Ziv et al., 2016). In agreement, vGSLs were found to act as signaling lipids to induce host programmed cell death (PCD) (Vardi et al., 2009; Liu et al., 2018). More recently, we further revealed that the host lipidome (both lipid content and composition)significantly changed in response to EhV infection(Zeng et al., 2019). Currently, there is still limited direct experimental evidence for the EhV genes proposed to play a role in sphingolipid biosynthesis.Therefore, it will be important to experimentally identify the viral genes involved in host sphingolipid metabolism, particularly given the proposed role for this sphingolipid in apoptosis and virus mediated sphingolipid metabolic regulatory mechanism.
Genetic transformation of eukaryotic cells is a valuable tool for the elucidation of gene functions and certain metabolic pathways, and allows us to study biochemical processes as well as viral infection mechanisms of the studied organisms. The ability to manipulate microalgae via genetic engineering in order to introduce or optimize desired traits will facilitate more extensive exploitation of these organisms since interest in the use of microalgae for research as well as commercial applications has increased in recent years (Hlavova et al., 2015; Xue et al., 2015; Velmurugan and Deka, 2018). However,information on transgene expression and genetic engineering for understanding molecular mechanisms and strain developments is very limited in coccolithophores (Endo et al., 2016).
The aim of this work is to design a novel vector construct for the transformation of the coccolithophore E. huxleyi and identify the possible function of EhVspt. Thus, we developed the methods of transient and stable transformation for E. huxleyi. The utility of glufosinate-ammonium selection marker ( bar) and green fluorescent protein reporter genes ( gfp) were examined for use in E. huxleyi. The genetic transformation was investigated using electroporation and the transformation conditions were determined with the gfp gene. The endogenous promoter and terminator of fucoxanthin chlorophyll a/c-binding protein gene “ fcp” were tested for the expression of bar and spt genes. The recombinant vector containing EhV-99B1- spt was transformed for a preliminary functional identification of viral spt gene.
2 MATERIAL AND METHOD
2.1 Cultivation of Emiliania huxleyi
Emiliania huxleyi BOF 92 strain, obtained from the Biology Department of Bergen University(Bergen, Norway) was grown in liquid 70% f/2-Si medium (Guillard, 1975) for usual cultivation. The culture conditions were 16±1 °C and the light regime was a 14-h꞉10-h light꞉dark cycle with about 50 μmol·photons/(m2·s) cool white fluorescent lights.The cultures were incubated with 2×104cells/mL.
2.2 Construction of expression vectors
All the genes fragments were obtained by PCR and the sequence of the primers and the PCR conditions were summarized in Table 1. Glufosinate-ammonium selection marker gene- bar (552 bp) was amplified by PCR from the commercial plasmid pCAMBIA3300(Biovector Science Lab, Inc.) as a Spe I/ Mlu I fragment using the primers A1/A2. The gene encoding green fluorescent protein- gfp (717 bp) was amplified by PCR from pGFP vector (Clontech) as a Spe I/EcoR I fragment using the primers A3/A4. As an attempt to increase gene transfer effi ciency and the transformant stability in E. huxleyi cells, constructscontaining larger fragments of promoter and terminator were considered. The endogenous fcpA promoter regions (FAP 484 bp and FBP 300 bp) of the fucoxanthin chlorophyll a/c-binding protein gene in E. huxleyi BOF92 were amplified by PCR as Bgl II/ Spe I and Not I/ Eco R I fragments respectively, with E. huxleyi genome as the template using the primer sets A5/A6 and A7/A8 respectively. Similarly, the fcpA terminator regions (FAT1 600 bp and FAT2 600 bp) were obtained as Mlu I/ Not I and Hind III/Xho I fragments by PCR using primers sets A9/A10 and A11/A12 respectively. Spt gene (2 613 bp) was amplified by PCR as Sma I/ Hind III fragment with E.huxleyi virus (EhV-99B1) genome as the template using the primer sets A13/A14. Premiers for both fcp promoters and terminators were designed based on the whole genomic sequences of E. huxleyi CCMP1516 (http://genome.jgi.doe.gov/Emihu1/Emihu1.home.html). Primers for spt were designed based on the genomic sequences of E. huxleyi virus 86 (EhV 86) (https://www.ncbi.nlm.nih.gov/nuccore/NC_007346). These products were cloned into the pMD19T (TaKaRa) vector respectively, for sequencing and analysis through tools from PLACE database (http://www.dna.aff rc.go.jp/PLACE/) and PlantCARE (http://bioniformatics.psb.ugent.be/webtools/plantcare/html/).
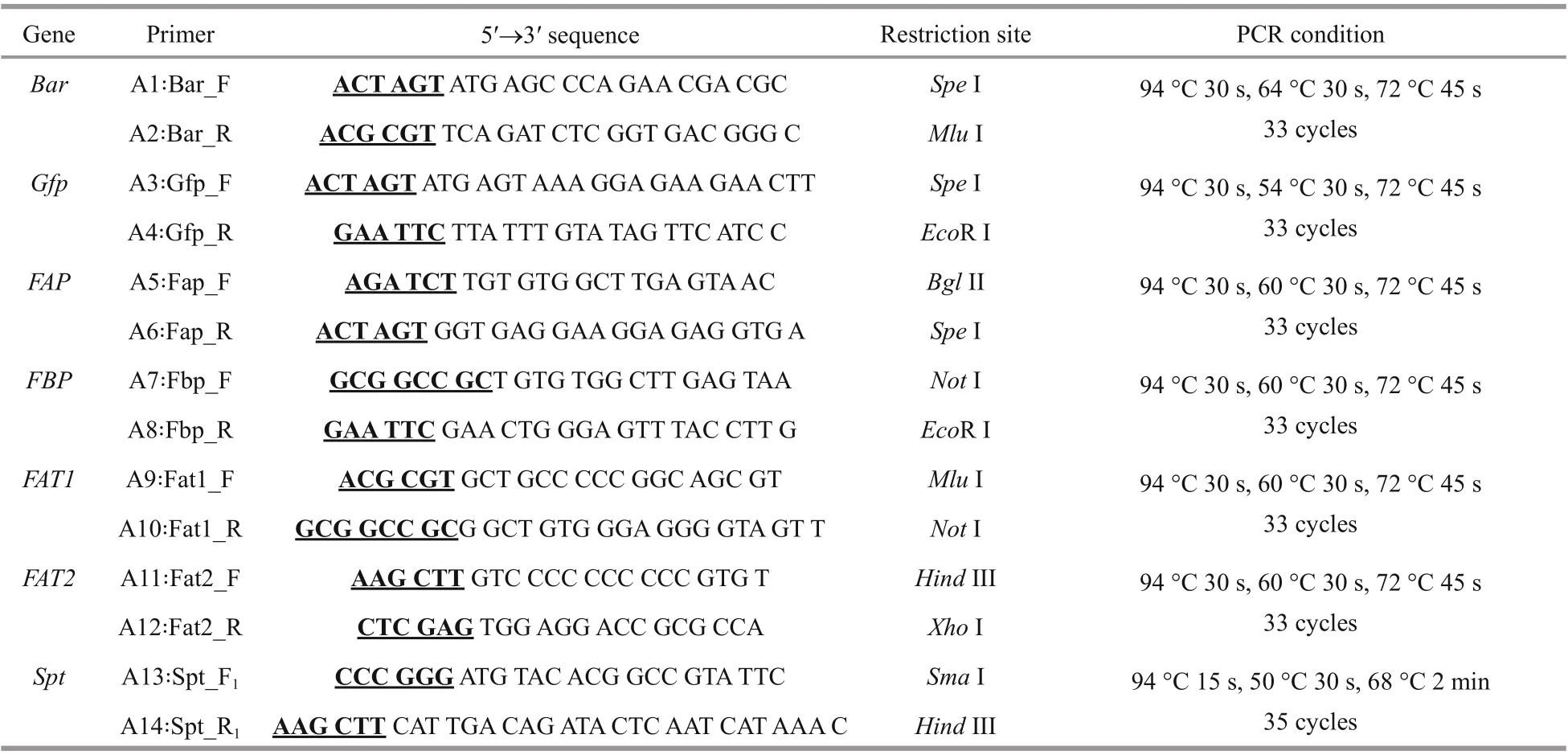
Table 1 Primers and PCR programs used in this study
The plasmid pSP73 (Promega Corporation,Madison, WI, USA) was used as the backbone transformation vector. Construction of the plasmids pEhux-I and pEhux-II used for transformation of E. huxleyi were shown as Fig.1a & b. Plasmid pEhux-I(Fig.1a) was constructed in several stages. The first step involved subcloning the 600 bp fcp terminator FAT2 into the Hind III/ Xho I sites of pSP73 to form the plasmid pSP73-FAT2. The genes of FAP ( fcp A promoter), bar, FAT1 ( fcp terminator) and FBP ( fcp A promoter) were ligated by Spe I, Mlu I, and Not I respectively as a Bgl II/ Eco R I fragment, which were inserted into pMD19-T vector for sequencing. The FAP/Bar/FAT1/FBP gene fusion (as a Bgl II / Eco R I fragment) was then linked into the Bgl II / Eco R I sites of pSP73-FAT2 to form the final basic construct pEhux-I (pSP73-FAP- bar-FAT1-FBP-FAT2). The pSP73 multiple cloning site (MCS) including Eco R I,Sma I, Xba I, and Hind III were preserved intact into which gene of spt was inserted as Sma I/ Hind III fragment. The recombinant vector harboring spt gene was designed as pEhux-I- spt (Fig.1c). The expression vector pEhux-II (Fig.1b), in which gfp is controlled by FAP promoter, was constructed by first inserting the 600 bp fcp terminator FAT2 gene (as a Hind III/ Xho I fragment) into the corresponding sites of pSP73. The FAP/ gfp gene fusion 1 201 bp (as a Bgl II/ Eco R I fragment) was then inserted into the Bgl II/Eco R I sites. The construct was designated pEhux-II(pSP73- FAP- gfp-FAT2).
2.3 Transformation of Emiliania huxleyi by electroporation
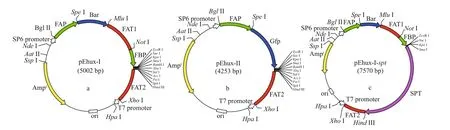
Fig.1 Construction of plasmids pEhux-I (a), pEhux-II (b), and pEhux-I- spt (c) used to co-transform Emiliania huxleyi BOF 92 strain
pEhux-I- spt bearing a marker gene “ bar” and pEhux-II carrying the reporter gene “ gfp” were cotransformed into E. huxleyi which should facilitate an identification of positively transformed clones.E. huxleyi cells were harvested at the mid-logarithmic phase (cell density of (1.5-2.0)×106cells/mL) by centrifuging (1 500× g, 5 min, 4 °C) and decalcified by suspension with 0.5-mol/L MES-NaOH buff er (pH 5.5) (Sekino and Shiraiwa, 1996) at 15 °C for 1 h.After the decalcification, cells were collected by centrifuging (1 500× g, 2 min, 4 °C) and suspended in 1-mL electric shock buff er (0.08-mol/L KCl,0.005-mol/L CaCl2, 0.2-mol/L mannitol, 0.2-mol/L sorbitol and 0.01-mol/L Hepes, pH 7.2) containing approximately 5.6×107cells. The created transformation vectors pEhux-I- spt and pEhux-II were re-suspended in distilled water and added to the above mentioned solution at the final concentration of 10 μg/mL. After incubation for 15 min at 4 °C without shaking, 300 μL of suspension was transferred into the electroporation chamber (Pulser/MicroPulser Cuvette, 0.4 cm gap; Bio-Rad Laboratories). The transformation of E. huxleyi was performed by electroporation using an electroporation system(Gemini SC, BTX, USA). The parameters, electrical field strength, used in electroporation was determined using the following equation: Critical electrical field strength E (V/cm)=10 000/(1.5×cell radius) (Čgovnik and Novaković, 2004). By observing electroporated cells under microscope to examine their mortality ratio, electroporation parameters were set with electrical field strengths of 1 500 V/cm and electrical shock time of 1.0-4.0 ms. To achieve high transformation effi ciency, the pulsed cells were kept in fresh 70% f/2-Si non-selective medium under constant illumination for 24 h to allow recovery and then glufosinate-ammonium (PPT) was added to these cultures at the final concentration of 50 μg/mL.The cultures were allowed to grow under standard culture conditions for one week before spreading on selection plates. Transformants were examined for gfp fluorescence by fluorescence microscope (Axio Vert A1, ZEISS, Germany).
2.4 Screening of transformants on solid medium
Growth experiments on solid media were performed in f/50 media supplemented with 1.5%agar (Sigma Aldrich) (Laguna et al., 2001). The media were autoclaved before adding 50 μg/mL PPT and ampicillin. Transformants cultures were collected by centrifugation at 1 300× g for 2 min and resuspended in 1-mL selective medium (~8×107cells). Transformed cells were screened on f/50 selective plates and were incubated photosynthetic conditions (14 h꞉10 h lightdark cycle) in upright position for the culture volume to integrate into the agar medium and then turned upside down for 2-3 weeks until the pigmented colonies appeared (wild type E. huxleyi as the control).
2.5 Southern hybridization
Transformants cultures re-growth experiments in liquid media were performed by scraping cells off the surface of the plates using disposable plastic inoculating rings and transferring them into liquid selective f/2 medium for 12 days under constant illumination. Genomic DNAs were extracted from transformed five strains (Nos. 1-5) following the method of hexadecyl-trimethyl-ammonium bromide(CTAB) (Ausubel et al., 1999) with some modifications. In briefly, algal cultures (250 mL) were collected by centrifugation at 1 300× g for 5 min at 4 °C. Cell pellets were suspended in 1-mL prewarmed lysis buff er (0.5% SDS, 20 μ g/mL proteinase K) and incubated at 55 °C for 30 min. Added 160- μ L 5-mol/L NaCl and 100- μ L pre-warmed 10% CTAB solution at 65 °C. After incubation at 65 °C for 10 min, the lysate was extracted with chloroform:isoamyl alcohol (24꞉1). The aqueous phase was collected and CsC1 was added to a final concentration of 1.2 g/mL, and ethidium bromide of 0.6 mg/mL.The solution was centrifuged at 200 000× g using a T-8100 rotor (Sorvall Discovery 100S centrifuge) for 6 h. The DNA band was removed by pipetting under UV irradiation and further purified using the ethanol precipitation method. Wild-type E. huxleyi genomic DNA was used as a negative control. 6.5 μ g of purified DNA were digested with Bgl II and transferred to a nylon membrane. The Biotinylated (dUTPs) spt specific probe was prepared by a random-prime method (Synbio Technologies) and hybridized with the membrane. Signals were detected by streptavidinhorseradish peroxidase conjugate and visualized using Chemiluminescence Detection kit (Advansta,WesternBrightTM ECL, USA).
2.6 Genomic PCR and qRT-PCR
Genomic PCR was performed using the DNA acquired above and PCR products were separated by electrophoresis in 1.0% agarose gel. Total RNA were isolated from transgenic E. huxleyi cells (1.84×105cells)using Plant RNA Isolation Reagent (Invitrogen Corporation, USA). The quality of the RNA sample was determined with a NanoDrop Spectrophotometer(ND-1000; Thermo Fisher Scientific Inc., USA). The first-strand cDNA was synthesised using the FastQuant RT kit (with gDNase) (TIANGEN, China). The cDNA were then subjected to qRT-PCR using standard methods to determine the expression level of spt gene in the transformed E. huxleyi cells. pEhux-I- spt vector(bearing spt gene) was used as the standard and decimal dilutions of plasmid pEhux-I- spt were tested to normalize the absolute expression level of spt gene in transgenic cells. Spt gene (140 bp) was amplified using the following primers: spt-f2: 5′-AGTCCGGTATCGTCTTGTCG-3′ and spt-r2: 5′-TACACCTTCAACCAAAACATAGA-3′.
A Thermo Scientific PikoReal PCR instrument(Thermo Fisher Scientific Inc., USA) and SYBR Premix Ex TaqTMII (TaKaRa) were used for qRT-PCR analysis. The assays were performed in a total volume of 10 μL containing 0.5 μL of the above-described cDNA, 0.1-μL SYBR Green I, 1-μL 10×PCR Buff er,0.8-μL dNTP (10 mmol/L), 0.4-pmol/L each of the 3′and 5′ primers, 0.5-U Taq HS, 3.4-mmol/L MgCl2,and RNase-free water. The qRT-PCR reactions were subjected to an initial denaturation step at 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s, 60 °C for 20 s and 72 °C for 20 s. A melting curve of reaction was used to determine the specificity of amplified products, which was obtained by performing a thermal cycle of 95 °C for 20 s, decreased to 60 °C for 1 min,increased again to 95 °C, with the temperature increase stepwise by 0.5 °C every 10 s. Templatefree, negative, pEhux-I- spt and single primers controls were established before the examination. All samples were analyzed separately, whereas triplicate Ctvalues of the same sample were averaged before drawing a standard curve, and the standard curve was used for statistical analysis.
2.7 Protein electrophoresis and Western blot
To examine spt protein expression, total protein was extracted from the transformants of E. huxleyi and wild type control using a Protein Extraction Kit(AR0102-10, Boster Biological Technology, USA)and protein concentration was determined using the Bio-Rad Protein Assay kit (Bio-Rad Laboratories).Proteins (40 μ g/well) were separated by SDS-PAGE(10% acrylamide) and electrotransferred onto nitrocellulose membranes (0.45 μm, ThermoFisher Scientific) for Western blot analysis.
Anti-LCB2polyclonal antibody (Abmart, Shanghai,China) was developed in rabbit using the recombinant EhV-99B1-LCB2(the catalytic subunit of EhV-99B1-spt) corresponding to the N-terminal region of spt (in 1꞉500 dilution). Protein bands were visualized using horseradish peroxidase conjugated goat anti-rabbit IgG as the secondary antibody (1꞉200 dilution) and a chemiluminescence detection system (Advansta,WesternBrightTM ECL, USA). The purified recombinant EhV-99B1-LCB2protein was used as a positive control.
2.8 Lipid extraction and thin-layer chromatography(TLC) analysis
Cultures in liquid were harvested by centrifugation at 1 300× g for 5 min at 4 °C. The dry powder of cell pellets were resuspended in 300 μL of methanol, and homogenized using the bead-based homogenizer(Tissuelyser-24, Shanghai Jingxin Industrial Development Co., Ltd., China) at 65 Hz for 2.5 min.Next, 600 μL of chloroform and 250 μL of Milli-Q water was added to the homogenate successively. The mixture was thoroughly vortexed after the solvent addition, followed by centrifugation at 12 000 r/min for 10 min at 8 °C to form a two-phase system. The down-layer was vacuum-dried in a Speedvac concentrator (Thermo Scientific, USA), and then was dissolved in 100 μL of methanol and separated by TLC on 50 mm×100 mm plates of silica gel GF 254(Qingdao, China) developed in the same direction with two diff erent solvent mixture of methyl acetate꞉isopropanol꞉chloroform꞉methanol꞉potassium chloride (0.9%) (25꞉25꞉25꞉10꞉9) and hexane꞉diethyl ether꞉glacial acetic acid (80꞉20꞉2), respectively. Spots in the TLC plates were visualized by exposing the plates to copper acetate (3%) in an oven for 10 min at 160 °C.
3 RESULT AND DISCUSSION
3.1 Vector elements construction
Transformation protocols require eff ective selection markers and reporter genes to discriminate successful transformants from untransformed cells.Genes that confer resistance to antibiotics or herbicide have been used successfully as selectable markers for marine algal transformants (Muto et al., 2013;Mussgnug, 2015). Previously, we chose ampicillin,kanamycin, streptomycin, G418, novobiocin,chloramphenicol, puromycin, and glufosinateammonium as selectable markers. In the resistant test,only PPT and G418 proved to be eff ective at killing E.huxleyi at low concentrations (25 μ g/mL) (data not shown here). The bar gene encodes for glufosinateammonium acetyltransferase that confers resistance to PPT. Since PPT has broad spectrum activity against bacteria, fungi and plants, it is useful as a selective agent for the construction of vectors in many organisms (Radakovits et al., 2010). In comparison to G418, PPT has some advantages, such as high activity,low toxicity, environmental friendly, and small side eff ects; therefore the bar gene is the most effi cient selective marker for E. huxleyi. The green fluorescent protein ( gfp) has been used as a universal reporter of gene expression and in subcellular localization analyses in various marine algae ( Miyagawa et al.,2009; Watanabe et al., 2011). The diatom fucoxanthinchlorophyll a/c binding protein gene ( FCP) promoter is eff ective in marine diatoms and other marine algae(Li et al., 2009; Miyagawa-Yamaguchi et al., 2011;Qin et al., 2012; Muto et al., 2013). Attempting to increase the transformation effi ciency and the transgene copy number, the larger fragments of 484-bp promoter/600-bp terminator (Fig.1a) and 300-bp promoter/600-bp terminator sequences of the fucoxanthin chlorophyll a/c-binding protein A gene( fcpA) in E. huxleyi BOF 92 were used to construct the cassettes (Fig.1b).
3.2 Development of transformation system for Emiliania huxleyi
Two general-purpose transformation vectors,pEhux-I and pEhux-II were constructed to facilitate effi cient introduction of heterologous genes in the E. huxleyi. pEhux-I harbored bar cassettes and MCS cassette. The primary selection for E. huxleyi cells harboring the vector was PPT resistance, encoded by the bar gene, which was flanked by the fcp A promoter(FAP) and the fcp A terminator (FAT1) (Fig.1a,Supplementary Fig.S1). pEhux-II contained the promoter fcp A (FAP) and terminator (FAT2) regions flanked the gfp gene and MCS to promote gfp gene expression eff ectively (Fig.1b, Supplementary Fig.S2). The promoter fcp A (FBP) and terminator (FAT2)regions flanked the MCS to promote effi cient expression of the inserted spt gene (Fig.1c,Supplementary Fig.S3). Several methods have been used in the transformation of algal cells, such as particle bombardment, electroporation, polyethylene glycol (PEG)-mediated transfer and agitation with glass beads or silicon fibers. Among these methods,electroporation has proven to be a powerful and economic method for the transient and stable expression of foreign genes in microalgae, including Chlamydomonas reinhardtii, Chlorella spp.,Dunaliella salina, Haematococcus pluvialis,Nannochloropsis sp., and Phaeodactylum tricornutum(Brown et al., 1991; Coll, 2006; Kilian et al., 2011;Niu et al., 2012, 2013). In this study, the binaryvectors pEhux-I- spt and pEhux-II were introduced into the E. huxleyi BOF92 simultaneously by electroporation. All the clones analyzed retained the non-selectable reporter gene under conditions selective only for the antibiotic resistance gene. It might be important to maintain selection pressure to retain bar activity and the stable expression of the non-selectable gene may therefore indicate that the two plasmids were integrated together at the same site in the genome, as most likely occurs in the moss Physcomitrella patens (Kammerer and Cove, 1996)and Phaeodactylum tricornutum (Falciatore et al.,1999). After being co-transformed by electroporation,E. huxleyi cells were incubated under 14 h꞉10 h lightdark cycling cultivation at 16 °C for 24 h and the gfp gene could be expressed in transformed E. huxleyi cells via fluorescence microscopy (Fig.2a & b). The culture was then supplemented carefully with 50 μg/mL of PPT and continued to culture. 25 μg/mL of PPT was suffi cient to completely inhibit the growth of E. huxleyi. Hence, 50-μg/mL PPT was used for selection of transformants. The negative controls were selected in the same fashion. After one week of culturing, the transformed E. huxleyi cells were allowed to grow on PPT selective medium, whereas none of the negative controls survived.
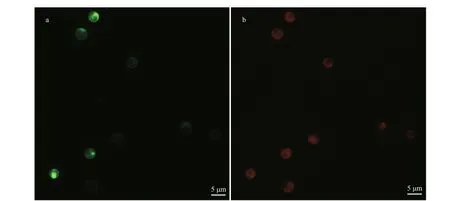
Fig.2 Microscopic images of Emiliania huxleyi BOF 92 strain cells transformed with pEhux-I- spt and pEhux-II vectors by electroporation and cultured for 24 h
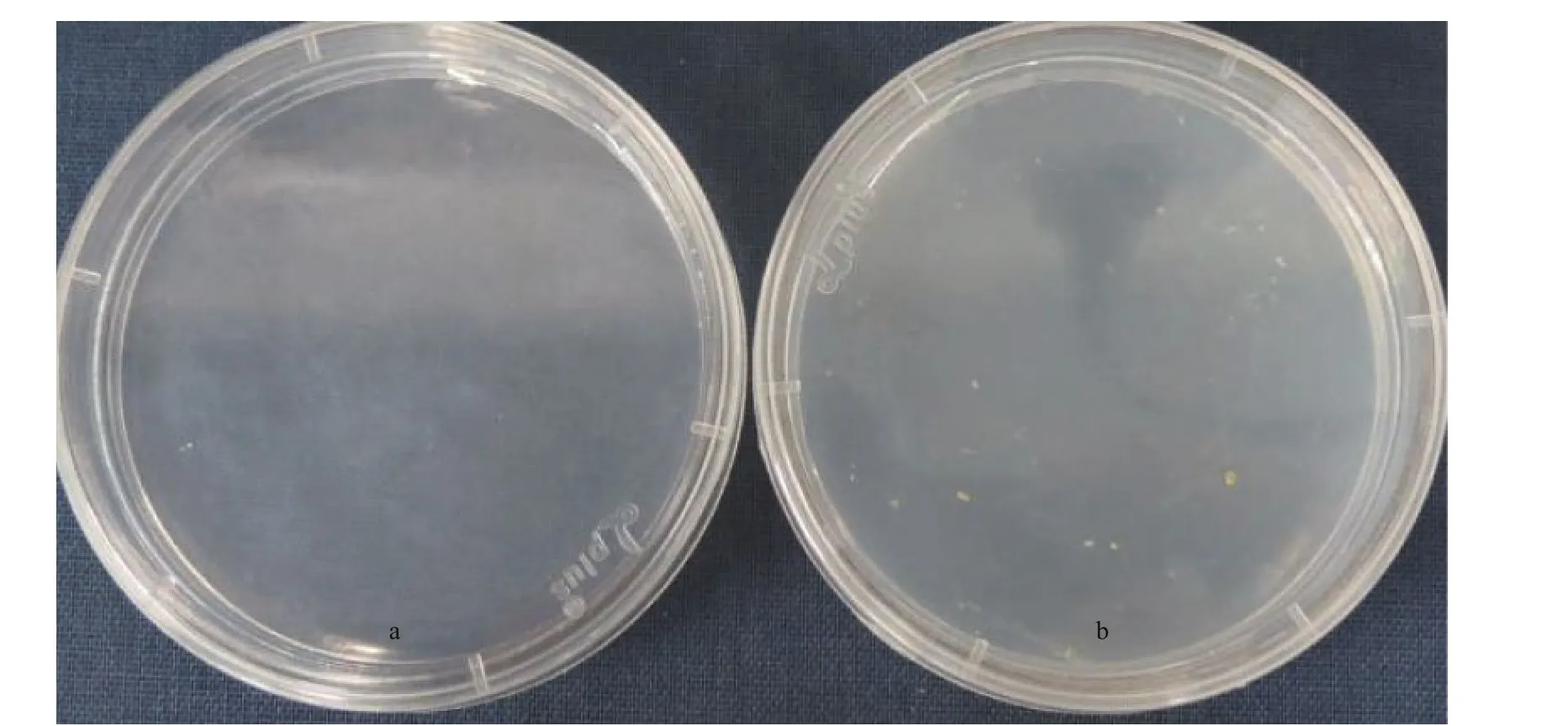
Fig.3 Cultures of wild-type (a) and transgenic (b) Emiliania huxleyi BOF 92 cells streaked on the glufosinate-ammonium containing medium (f/50 medium) cultured for 20 days after electroporation
3.3 Stability of the transformed phenotype
Transformants cultures in liquid selective medium were collected by centrifugation and then screened on f/50 selective plates containing 50- μ g/mL PPT. After for 4-5 days cultivated, some very small single colonies could be observed and the pigmented colonies appeared after extended incubation about three weeks (Fig.3a & b).
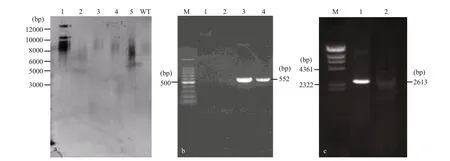
Fig.4 Southern blot and genome PCR analysis of wild-type and transformants of Emiliania huxleyi BOF92
The survived cells on selection plates were scraped off using disposable plastic inoculating rings and transferred into liquid f/2-Si selective medium,meanwhile untransformed cells inoculated in parallel.After one-week cultivation, transformed cells from solid media re-grown slowly, while untransformed cells inoculated in paralleled could not grow even after 2 weeks, indicating that bar was successfully expressed and resulted in PPT resistance, indicating that a nuclear transformation system has been developed for E. huxleyi BOF92 using electroporation to introduce the selectable marker bar gene into cells.Transformants were obtained when the fcp promoter was used to drive bar expression and a numbers of transformants were generated (3.8 colonies/106cells).It showed that cultures have to be adapted to permanent light for an extended period as done with P. tricornutum since gene expression is apparently regulated in a circadian rhythm (Oeltjen et al., 2004).Therefore, we could infer that fcp promoter has higher effi ciency under continuous light conditions. The pulsed E. huxleyi were kept in nonselective medium for 24 h to allow recovery before spreading on selection plates. In the present study, the transformation effi ciency (3.8 colonies/106cells) was lower than those of the results reported for P. tricornutum transformation systems by using electroporation(1 colony/105cells) (Niu et al., 2012) and microparticle bombardment (6.5 colonies/106cells) (Miyagawa et al., 2009). The first stable nuclear transformation of coccolithophore, Pleurochrysis carterae was established by means of polyethylene glycol (PEG)-mediated transfer and acquired a transformation effi ciency approximately of 9.5 colonies/106cells(Endo et al., 2016). Our experiment was conducted on constructs containing larger fragments of the fcpA promoters FAP (600 bp)/FBP (300 bp) and the fcpA terminator FAT1/FAT2 (a 600-bp region from the end of the coding region), though it seemed that the use of these constructs did not acquire the higher transformation effi ciency. Electroporation commonly results in highly variable integrated transgene copy numbers and low copy transformants. In electroporation, electric pulses are varied in intensity,duration and number to control the effi ciency of exogenous DNA delivery (Qin et al., 2012). In short,the results of this study demonstrate that electroporation might be an eff ective and a convenient method for the transformation of coccolithophore E.huxleyi, though further analysis should be performed to confirm a clear relationship between transformation effi ciencies and electroporation conditions in E.huxleyi. The transformation method has to be optimized to attain higher transformation effi ciencies.
3.4 Analysis of the stable co-transformants
The stable integration of the spt into the genomic DNA of E. huxleyi was demonstrated by Southern blot and genome PCR. Southern hybridization was performed with a probe with a 625-bp fragment of spt. The genomic DNA isolated from a transgenic strain was digested with Bgl II. Southern hybridization indicated that approximately 3-4 spt sequences were integrated into the genomic DNA of two transformed cells (Fig.4a). PCR screening acquired a 552-bp bar gene (Fig.4b) and an expected 2 613-bp band of spt gene (Fig.4c) in the transgenic cell lines using primers spt-F1/ spt-R1and bar-F/ bar-R, respectively, while absent in wild-type. Fragment obtained from transformants were sequenced to confirm that the fragment was amplified from the introduced gene.Based on these results, we concluded that relatively stable transformation of spt was achieved.
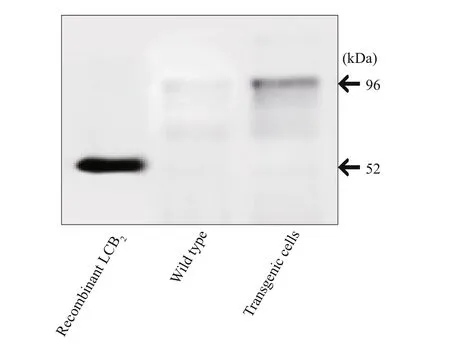
Fig.5 Western blot analysis
The copy number of the EhV-99B1- spt gene was evaluated by absolute qRT-PCR assay. A DNA melt curve analysis was implemented with specific primers spt-f2/ spt-r2and E. huxleyi cDNA. The pEhux-I- spt as detection template was carried out to verify amplification specificity. In order to detect the quantity of RNA in transformation group and wild type E.huxleyi cells, decimal dilutions of pEhux-I- spt vector were tested, and their Ctvalues linearly related to the logarithm of the starting cDNA copy number were plotted as the standard curve of the reaction(Supplementary Fig.S4). The parameters obtained for the SYBR Green I qRT-PCR: the standard curve y=-3.427 67 x+39.468 41, correlation coeffi cient ( R2)0.998 and percentage effi ciency (EFF) 95.7%. In the transformation group E. huxleyi, the absolute expression level of spt gene was analyzed based on the amplification curves and melt plots and obtained 28.21 of Ctvalue. By calculation, copy number of the spt gene was approximately 2.06 copies/cell in the transgenic E. huxleyi cells, suggesting that these genes had been integrated successfully into the genome of E. huxleyi and were transcribed in transformed cells under the control of the endogenous fcp putative promoter. In transgenic organisms, the transgene copy number can greatly influence the expression level and genetic stability of the target gene, and thus the estimation of transgene copy number is most important. Comparative experiments were also conducted on constructs containing only medium size fragments of the fcp A promoter (484 bp),which resulted in the lower transgene copy number of 1.67 copies/cell (data not shown). Use of the larger size both of the fcp promoter/terminator fragments might be needed to acquire stable insertion into genome.
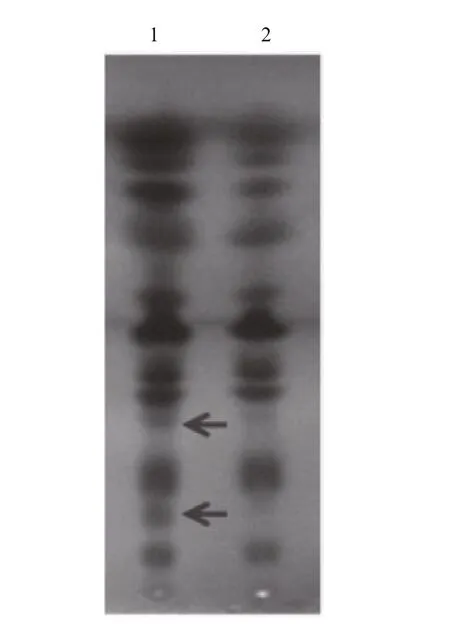
Fig.6 Thin-layer chromatography (TLC) of the total lipids in Emiliania huxleyi BOF 92 exposing the plates to copper acetate (3%)
The Western blot analyses were performed to evaluate expression of the spt gene in E. huxleyi transgenic cells using polyclonal antibodies specific for EhV-99B1-LCB2. As determined by immunoblots,the LCB2protein (~52.0 kDa) was present in the transformed cell lines examined while absent in wildtype cells (Fig.5). In this work, a successful growth of transformants could be kept and growing on solid selective media and the integration of spt and bar genes into the nuclear genome of E. huxleyi appeared to be stable.
3.5 Changes in total lipid by TLC analysis
SPT is the key enzyme in sphingolipid biosynthesis and it is considered to be a heterodimer of two subunits of Sptlc1(LCB1) and Sptlc2(LCB2) (Hanada et al., 2000). Interestingly, EhV-99B1- spt presented a single open reading frame (ORF), in which its N-terminal domain most closely resembled the LCB2subunit and the C-terminal domain most closely resembled the LCB1subunit of eukaryotic SPT(Wilson et al., 2005; Liu et al., 2012). To evaluate the possible function and activity of EhV-99B1- spt, we developed the new vector system for heterologous gene expression in E. huxleyi, producing transformants with EhV- spt gene. TLC analysis result clearly showed that a significant change of total lipid compositions in transformed E. huxleyi cells (Fig.6),indicating that the EhV- spt had certain catalytic activity.
4 CONCLUSION
We have successfully transformed the constructs incorporating a tandem cassette containing bar, spt,and MCS driven by the fcpA promoter/ fcpA terminator of E. huxleyi BOF92. The spt gene was integrated successfully into the nuclear genome and expressed in the E. huxleyi cells, which revealed the eff ectiveness of general transformation vector. For a functional identification, EhV-99B1- spt gene expression resulted in a clearly change of total lipid compositions in transformed E. huxleyi cells. The creation of a transformation system for E. huxleyi provided additional genetic resource with potential for exploring basic biological questions such as E. huxleyi virus-host interaction and also might make the organism a potential bioreactor of bioactive metabolites.
5 DATA AVAILABILITY STATEMENT
The datasets generated during and/or analyzed in this study are available from the corresponding author upon reasonable request.
6 ACKNOWLEDGMENT
Our deepest thanks go to Prof. Gunnar BRATBAK(Department of Biology, University of Bergen) for providing the E. huxleyi BOF92 strain and the E.huxleyi virus 99B1 strain friendly. We also would appreciate Prof. Kehou PAN and Baohua ZHU (Ocean University of China) for providing plasmid pSP73.
杂志排行

Journal of Oceanology and Limnology的其它文章
- Predicting sediment flux from continental shelfislands,southeastern China*
- Laboratory simulation of dissolved oxygen reduction and ammonia nitrogen generation in the decay stage of harmful algae bloom*
- Development of high-resolution chloroplast markers for intraspecific phylogeographic studies of Phaeocystis globosa*
- Effects ofiron and humic acid on competition between Microcystis aeruginosa and Scenedesmus obliquus revealed by HPLC analysis of pigments*
- Effect of river plume on phytoplankton community structure in Zhujiang River estuary*
- Exploring the sublethal genotoxic effects of class II organophosphorus insecticide quinalphos on freshwater fish Cyprinus carpio