A Zinc(II) coordination complex containing multi-benzimidazole ligand: synthesis, characterization and theoretical research
2021-04-10ZHOUHongbinLIJiXIAOFengpingMENGXianggao
ZHOU Hongbin, LI Ji, XIAO Fengping, MENG Xianggao*
(1.College of Chemistry, Central China Normal University, Wuhan 430079, China;2.School of Biological and Environmental Engineering, Guiyang University, Guiyang 550005, China)
Abstract: One new multi-benzimidazole (bzim) zinc (II) coordination compound, [Zn(NTB)Cl]2·(ZnCl4)·4(CH3OH) (1) (NTB=Tris(2-benzimidazolylmethyl)amine), has been synthesized and characterized by infrared absorption spectroscopy, elemental analysis and X-ray diffraction. Compound (1) was crystallized in the triclinic P-1 space group with a=1.3874(2) nm,b=1.4095(1) nm,c=1.6214(2) nm;α=86.440(3)°,β=87.296(2) °,γ=70.699(3) °,V=2.9855(6) nm3; μ=1.520 mm-1,ρc=1.504 mg·m-3,F(000)=1384,R1=0.0777, and wR2=0.2068 for 9 770 observed reflections (I > 2σ(I)). Two [Zn(NTB)Cl]+ cationic units, one (ZnCl4)2- anionic unit and four methanol solvent molecules are existing in its asymmetric unit. The component ions are linked by N—H…O, N—H…Cl, O—H…Cl, C—H…π and π…π interactions into a complex three-dimensional network. A quantitative analysis of the intermolecular interactions in crystal (1) were analyzed by using Hirshfeld surface and fingerprint plot, indicating that the strong spikes are mainly ascribed to H…O and H…Cl interactions. The electronic structure was theoretically calculated at the DFT(RB3LYP)/6-31G+(d, p) level for one discrete [Zn(NTB)Cl]+ unit. Frontier molecular orbital analysis shows that the less delocalization between these BD orbits of CH2 group and BD* orbits of benzimidazole group may be facilitating for the formation of the Δ and Λ configurations for the NTB ligand. The mulliken charge analysis shows that the three nitrogen-bonded hydrogen atoms (H3/H5/H7) in the [Zn(NTB)Cl]+ units possesses higher positive charges(about 0.28 a.u.) which can facilitate the formation of N—H…X interactions.
Key words: multi-benzimidazole; Zinc (II) coordination compound; crystal structure; Hirshfeld surface; DFT
Carbonic anhydrases (CAs) are widespread zinc-containing enzymes in plants and animals which can catalyze a crucially physiological reaction: the hydration of carbon dioxide to bicarbonate and protons (Fig.1)[1]. The active site of CAs are unambiguously known as three histidine imidazole groups and a water solvent molecule (Fig.2), forming a tetrahedral coordination configuration around the central Zn(II) atom[2]. In order to ascertain its catalytic mechanism, many studies have been mainly focused on the structural mimicking of the active sites of CA. In recent years, multi-benzimidazole ligand tris(2-benzimidazolylmethyl)amine (NTB) has been often used in the design and synthesis of metal-containing enzymatic-mimicking compounds due to its structurally similar to the histidine imidazole group[3-5]. Herein, we used NTB and ZnCl2as the starting materials and obtained a mononuclear Zn(II) compound under a certain conditions. In this paper, its structural characteristic, Hirshfeld surface and frontier orbits have been reported and discussed which can be useful for the future structure-activity-relationship research about CAs.

Fig.1 Simplified scheme for the catalytic mechanism of carbon anhydrase activity

Fig.2 Schematic representation of the active site of carbonic anhydrase.
1 Experimental
1.1 Materials and measurements
All chemicals were of reagent grade as received from commercial sources and used without further purification. C, H, N elemental analyses were performed on an Elementar Vario MICRO E III analyzer. Ultraviolet-visible (UV-vis) spectra was recorded on a CARY 60 spectrometer. Infrared (IR) spectra was recorded as KBr pellets on a Perkin Elmer spectrometer (4 000-400 cm-1). All calculations were made by Gaussian 09 software[6]and Gauss View 5 software was used to visualize the optimized structures[7]. The optimized molecular structure parameters of both the compounds are calculated through density functional theory (DFT) method. And the ground state optimizations in the gas phase were carried out using the crystallographic coordinates as the starting point with the method of RB3LYP/6-31G+(d, p). Crystal Explorer 17.5 program was used to perform the Hirshfeld surfaces analysis[8].
1.2 Synthesis of compound (1)
Tris(2-benzimidazolylmethyl)amine (NTB) was prepared according a earlier reported procedure[9]. NTB (0.204 g, 5.010-4mol) was dissolved in hot methanol (30.0 mL) and added to a solution of ZnCl2(0.102 g, 7.510-4mol) in 30.0 ml of methanol. Crystals were obtained by slow evaporation of a methanol solution. The mixture was stirred for 30 min at 50 ℃, and then the reaction mixture was filtered. Colorless block crystals suitable for X-ray study were obtained at the bottom of the vessel after the filtrate solution was evaporated at room temperature for two weeks (0.270 g, yield: 67%); Element Analysis: calcd.: C 44.76%, H 4.19%, N 14.05%; Found: C 44.90%, H 3.97%, N 14.25%; UV-Vis: 277.2 nm (ε=1.28104L·cm-1·mol-1). IR: 3 409 (m), 1 623 (w), 1 549 (m), 1 473(s), 1 451(s), 1 144(m), 1 111(s), 1 088(s), 752(s), 627(s).
1.3 X-ray crystallography
A colorless crystal with dimension of 0.12 × 0.12 × 0.10 mm3was selected for X-ray data collection on a Bruker SMART APEX CCD area-detector diffractometer equipped with a graphite-monochromatic Mo-Kαradiation (λ=0.071073 nm) at 100(2) K. A total of 10 604 reflections were collected in the range of 1.533°≤ λ ≤ 25.250°, of which 9 770 independent reflections (Rint=0.0389) were collected. In the data reduction, Lorentz and polarization effects and empirical absorption corrections were applied[10](Tab.1). The structure was solved by direct methods and refined by full-matrix least square techniques based on F2using the SHELXS-97 and SHELXL-2014[11-12]programs, respectively. All non-hydrogen atoms were refined anisotropically. H atoms bonded to N atoms were located in difference Fourier maps and refined with restraints N—H=0.086(2) nm and Uiso(H)=1.2Ueq(N), O—H=0.082(2) nm and Uiso(H)=1.5Ueq(O), respectively. All the remaining H atoms were positioned geometrically and treated as riding modes with d(C—H)=0.093 nm, Uiso(H)=1.2Ueq(C, aromatic) and d(C-H)=0.097 nm, Uiso(H)=1.2Ueq(C, methylene).Some selected bond lengths are given in Tab.2. CCDC: 1947285. The crystal data can be obtained from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/ cif or from the authors.

Tab.1 Selected crystallographic data of compound (1)
2 Results and discussion
2.1 Crystal structure description
For compound (1), it was crystallized in the triclinicP-1 space group and its asymmetric unit was composed of two [Zn(NTB)Cl]+cations, one (ZnCl4)2-anion and four methanol solvent molecules. A very similar analog of (1) was reported crystallized in the monoclinic C2/c space group with the formula of [(NTB(Zn)Cl)]2(ZnCl4)2-(EtOH)4[13]. In both the [Zn(NTB)Cl]+moieties, the zinc(II) ions are coordinated with a N4Cl donor set with the τ parameters of 0.51 (1) for Zn1 and 0.52 (1) for Zn2, respectively, indicating slightly distorted trigonal bi-pyramidal polyhedrons (Fig.3). Their base planes are formed both by three benzimidazole (bzim) nitrogen atoms and the two axial sites are occupied by one tertiary nitrogen atom and one chloride anion. The bond lengths Zn—Nbzimare in a range of 0.201 7(6) nm to 0.205 6 (6) nm. The bond distances between Zn(II) and the tertiary nitrogen atoms (N1 and N1A) are 0.252 9(7) nm and 0.253 9(7) nm, which are much longer than the Zn—Nbzimdistances (Tab.2). The significant elongation is frequently observed in some Zn(NTB)-containing analogs[14-15]. When the weak coordination bonds were omitted, the Zn1 and Zn2 atoms can be also regarded as a tetrahedral configuration. The Zn—Cl bonds in the two cationic and anionic ions are comparable with each other, ranging from 0.225 1(2) nm to 0.230 2(2) nm. The distortion of the trigonal bipyramidal coordination environments around Zn(II) centers are indicated by the deviations of the equatorial angles from 120°. For Zn1 and Zn1A, these three angles are 113.1(3)°/110.8(3)°/115.1(3)° and 113.7(3)°/110.0(2)°/114.7(2)°, respectively. They deviate by 0.055 0 and 0.055 8 nm away from their respective basal planes. For the tetrahedral ZnCl4anion, the four bond distances and angles are similar to some analogs (Tab.2-Tab.3)[13].
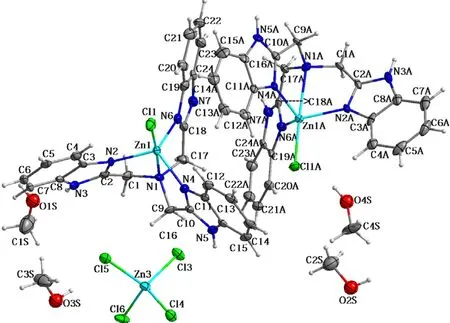
Fig.3 ORTEP molecular structure of compound (1) shown as 30% thermal ellipsoid probability
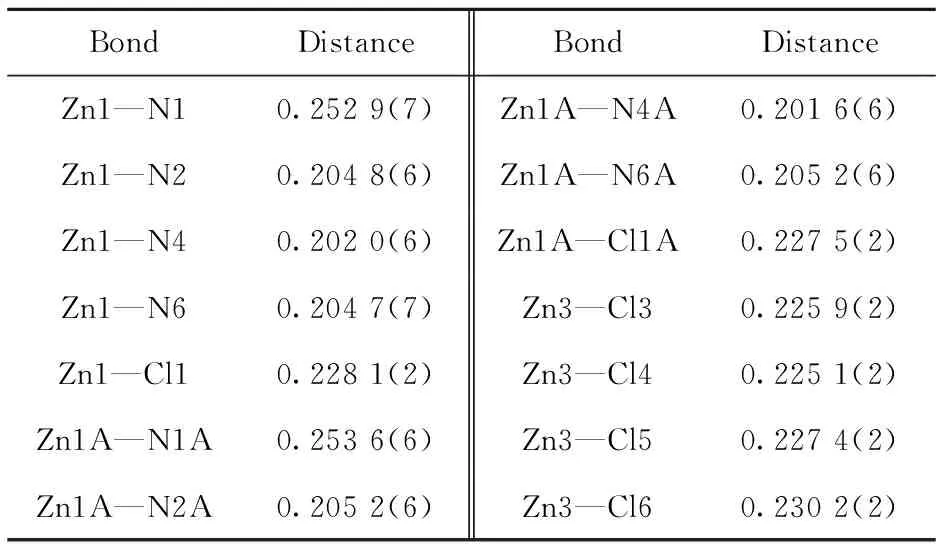
Tab.2 Selected bond distances for compound (1) nm
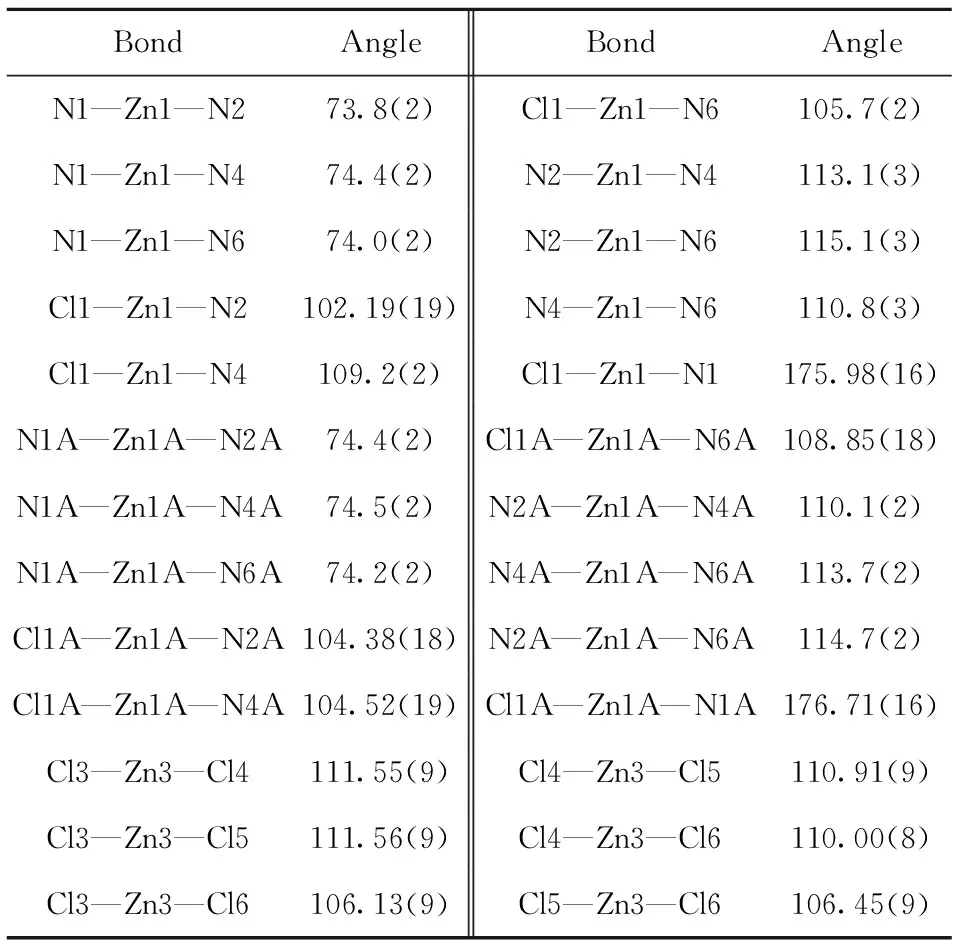
Tab.3 Selected bond angles for compound (1) °
From a view along the Namine—Zinc—Cl axial direction, there exist the Δ and Λ configurations for the N—(CH2)3—moiety in NTB ligand, which can assist for the construction of some an-centric supra-molecular structures[16]. In the crystal of compound (1), there are equal amounts of Δ and Λ configurations of [Zn(NTB)Cl]+cations due to the inversion center inP-1 space group. Further research can be focused on the effect of metal ions, solvent, temperature etc. on the assembling of a specific Δ or Λ configuration (Fig.4).
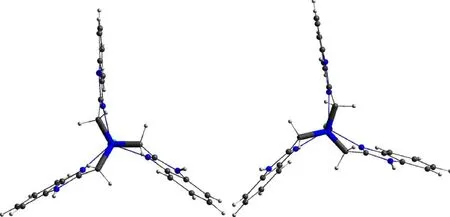
Fig.4 Representation of a pair of chiral conformers corresponding to clockwise (left, Δ) and anticlockwise (right, Λ) conformations of the tripodal ligand in the [Cu(NTB)]+ cation
In the crystal packing of (1), the component ions are linked into complex three-dimensional network by combination of intermolecular N—H…Cl, N—H…O and O—H…Cl hydrogen bonds (Fig.5 and Tab.4). Further analysis by using Platon indicates that C—H…π and π…π interactions are extensively occurred between symmetric benzimidazole groups with the nearest centroid-to-centroid distance being only 0.343 4(2) nm[17].

Fig.5 Part of the three-dimensional hydrogen-bonded network in compound (1) by N—H…O/Cl, O—H…Cl hydrogen bonds and π…π interactions shown as colored dashed lines

Tab.4 Hydrogen bond parameters in compound (1)
2.2 Hirshfeld Surface
To quantitatively classify the intermolecular actions between two types of elements in a crystal packing, three molecular Hirshfeld surface analysis for the three Zn1-, Zn2- and Zn3-containing moieties have been conducted by using the Crystal-Explorer software[8,18](Fig.6(a)-6(c)).
From Fig.6(a) and Fig.6(b), one can easily found that the two cationic [Zn(NTB)Cl]+units have similar Hirshfeld surfaces mapped over dnorm. Their full fingerprints are also similar to each other when these distinct spikes are compared (Fig.6(d)-6(e)). For instance, each Zn1-containing cationic unit constitutes two N—H…O and one N—H…Cl hydrogen-bonds as an electron-donor. The H…O and H…Cl interactions occupy about 4.1% and 7.2% with the distinct spikes residing at the upper-left corner of the fingerprint plot, respectively. The other two clear spikes are the H…C (23.8%) contact which can be regarded as the C—H…π interactions. For the π…π interactions, it can also be discriminated from the central region of the fingerprint plot with a reciprocal contact surface of 8.2% for C…C/N element contacts. Although the largest surface area is dominated by the H…H contacts (45.7%) indicating the existence of van der Waals’ interactions, they are not dominant driving forces in the crystal packing due to the lack of short distanced spikes. For the Zn1A-containing coordination unit, these H…O, H…Cl, H…C, C…C/N and H…H contact surface areas are 4.3%, 11.9%, 24.2%, 7.2%, 42.8%, respectively, indicating similar intermolecular interactions. As for the tetrahedral configured [ZnCl4]2-unit, its intermolecular interaction is only the Cl…H contacts with a fingerprint area of 94.9% (Fig.6(f)), indicating the formation of strong N—H…Cl hydrogen bonds. In one word, from the Hirshfeld surface analysis the intermolecular interactions can be quantitatively analyzed according a structural research need.
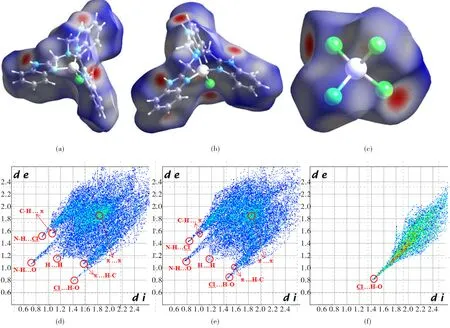
Fig.6 Hirshfeld surfaces mapped with dnorm for the Zn1 (a), Zn1A (b) and Zn3-containing coordination ions (a)-(c) and their full fingerprint plots (d)-(f)
2.3 Quantum mechanical study
2.3.1 Stability The optimization of one [Zn(NTB)Cl]+unit within the Cipoint group is carried out.The final outcome gives a better convergence criterion and no imaginary frequency has been observed. The total energy for it is -1 836.299 097 a.u. and the energies of HOMO and LUMO orbits are -0.338 02 and -0.151 48 a.u., respectively. The HOMO-LUMO gap is 0.186 54 a.u. and the dipole moment is 13.688 6 D. A narrow frontier bond gap may be helpful for its spectroscopic and biological activity.

2.3.2 Frontier molecular orbital composition In order to ascertain the structure and bond characteristics in [Zn(NTB)Cl]+unit, these highest occupied and lowest unoccupied frontier molecular orbital populations have been analyzed by us. The atomic orbital composition from different type of atoms in the frontier molecular orbits were expressed as the atomic orbital coefficient square sum in the type of atomic orbits and corrected by normalizing the specific molecule orbital[19]. These atoms in [Zn(NTB)Cl]+can be classified into seven moieties according to their coordination environment and connectivity: 1) N2/N3-containing benzmidazole group; 2) N4/N5-containing benzmidazole group; 3) N6/N7-containing benzmidazole group; 4) the three methylene groups; 5) amine N atom; 6) chloride Cl1 atom; 7) metal Zn1 atom.
According to an orbital contribution analysis (Tab.5), it can be seen that the compositions of HOMO and LUMO orbits are almost averagely coming from three benzimidazole groups, ~32% for the former and ~24% for the latter, respectively (Fig.7). But in the LUMO orbit the three —CH2— groups and the central Zn1 atom also provide apparent contribution to the LUMO orbit (9.0% and 11.9%). The distributions of the HOMOs and LUMOs also demonstrate that some specific electron transferring may occur between these benzimidazole groups. In the UV-vis spectrographic determination, the absorption of π…π electron transfer at about 277.2 nm may be also related with these frontier molecular orbits. In the other frontier molecular orbits, it’s found that the three methylene groups are all giving more contributions than that in the HOMO and LUMO orbits. These results here demonstrate that the electrons are mainly delocalized into three benzimidazole groups.

Fig.7 The frontier molecular orbits from HOMO-4 to LUMO+4 in compound (1)
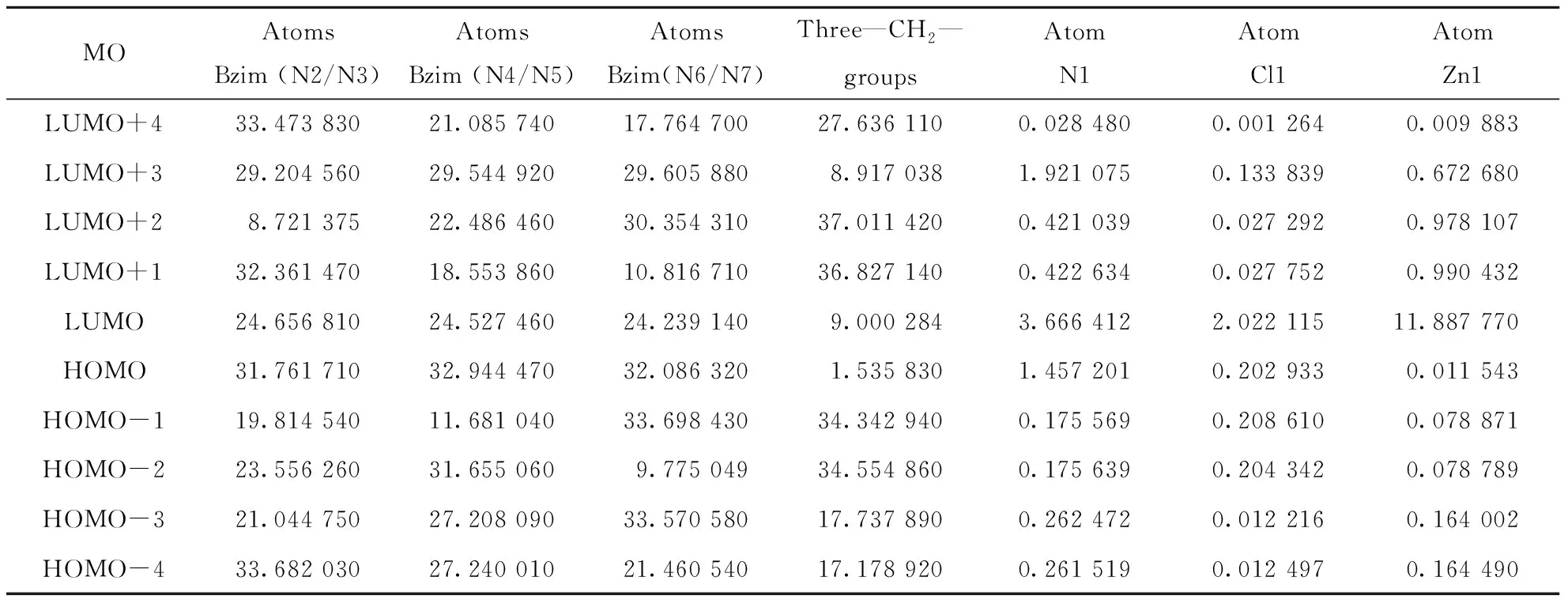
Tab.5 Frontier molecular orbital (MO) compositions from different type of groups and atoms in (1) %
As for the mulliken charge, it can be easily seen that all the three benzimidazole N atoms (N2/N4/N6) have carried more positive charges (Tab.6), indicating the existence of more stronger interactions between the nitrogen atoms and metal Zn1 center. However, the axial amine N1 and chloride Cl1 atoms possess more negative charges (-0.34 a.u. and-0.45 a.u.). Thus, it can be concluded that the coordination bonds between the Zn—Nbzimshould be stronger than that of the Zn—Namineand Zn—Cl1. Additionally, the H3, H5 and H7 atoms are easily forming hydrogen-bonding because of their more positive charges (Tab.6) which is also verified by the crystal packing analysis.
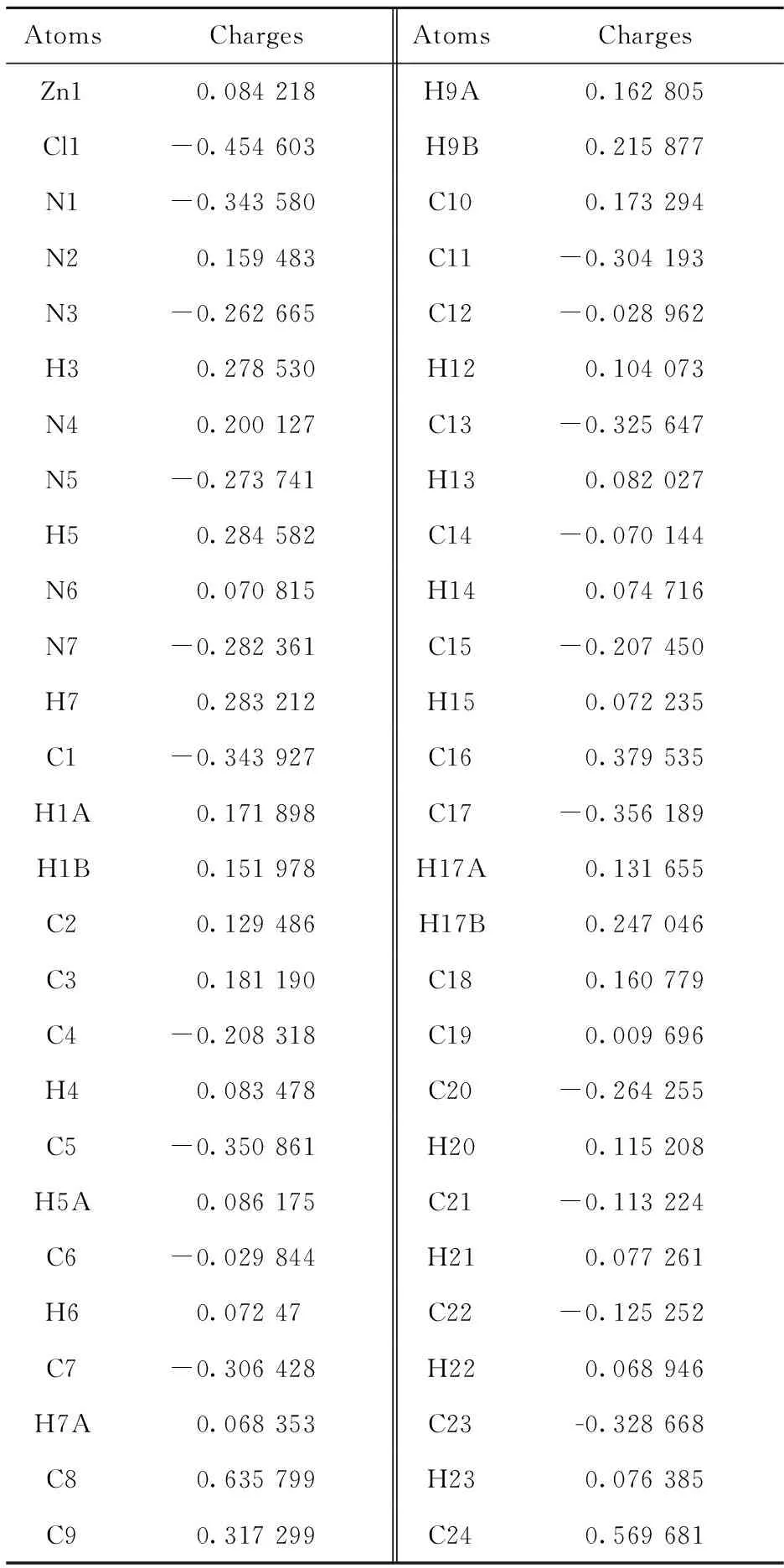
Tab.6 Mulliken charges in the optimized structure of compound (1) a.u.
From Tab.7, we can get some selected interactions between the electron-donors and the electron-acceptors orbits. The degree of their interactions were representing by the stabilization energies (E2), as is well known that the larger the energy (E2) is, the stronger the interaction between the orbits (iandj) is the more tendency of orbital i providing electrons to orbitalj. And more electron-delocalization is formed in the studied bonds regions. In Tab.7, it can be easily seen that these three Namine—C and their neighboring C—C bonding orbits are all giving little stabilization energy (2.78 and 2.14 kJ·mol-1, respectively.) with the C=N double bonds in the imidazole groups. Thus, the less delocalization between these bonding and anti-bonding orbits may be helpful for the formation of the Δ and Λ configurations around the amine N atoms. For the central Zinc(II) coordination bonds, they form the different the stabilization energies between the four LP N orbits and one LP Cl1 orbital with the LP×Zn1 orbital (Tab.7).The weakest one is only 11.54 kJ·mol-1which can be also elucidating the apparently longest Zn-N bond (0.270 8(1) nm) among the coordination bonds.

Tab.7 Selected calculated results of the compound (1) at the B3LYP/6-31G level by NBO analysis
3 Conclusion
In summary, we have reported a new zinc (II) coordination complex, [Zn(NTB)Cl]2·ZnCl4·4(CH3OH), in which the component ions are linked into a complex three-dimensional network by a combination of N—H…O, N—H…Cl and O—H…Cl hydrogen bonds. C—H…π and π…π interactions are also observed in the crystal packing. Hirshfeld surface, frontier molecular orbits compositions and natural bond orbital analysis were also performed for a [Zn(NTB)Cl]+unit in details. The theoretical analysis of this compound is believed to be helpful and guiding for the spectroscopic and biological enzymatic mimic research in the future.
