Harnessing CRISPR-Cas system diversity for gene editing technologies
2021-04-09AlexanderMcKayGaetanBurgio
Alexander McKay, Gaetan Burgio
Department of Immunology and Infectious Diseases, John Curtin School of Medical Research, Australian National University, Canberra, ACT 2601, Australia.
Abstract The discovery and utilization of RNA-guided surveillance complexes, such as CRISPR-Cas9, for sequencespecific DNA or RNA cleavage, has revolutionised the process of gene modification or knockdown. To optimise the use of this technology, an exploratory race has ensued to discover or develop new RNA-guided endonucleases with the most flexible sequence targeting requirements, coupled with high cleavage efficacy and specificity. Here we review the constraints of existing gene editing and assess the merits of exploiting the diversity of CRISPR-Cas effectors as a methodology for surmounting these limitations.
Keywords: CRISPR-Cas systems, gene editing, biological evolution, DNA repair, classification, DNA transposable elements
Introduction
Gene editing is a cornerstone technology for the production of genetically modified organisms with diverse applications spanning the research, medical and pharmaceutical fields. Current approaches to sitespecifically edit genomes rely on hijacking the host cell DNA repair pathways responsible for fixing double strand breaks (DSBs) and modify or completely inhibit the function of a target gene[1-2]. In concert with a delivery strategy, such as transfection or electroporation[3-6], a programmable endonuclease is introduced into the host cell, and generates a single or a double-stranded break (DSB) in the DNA[1,7]. A revolution in gene editing in recent years has precipitated as a result of the discovery that a ribonucleoprotein complex called CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats-CRISPR associated protein) can be programmed to specifically target loci in the genome of an organism[1].
To further improve upon the properties of the CRISPR-Cas9 gene editing system, and understand the ecological role of the technology, a significant focus has been exploring the evolutionary diversity of CRISPR-Cas systems. The CRISPR-Cas9 editing complexes were originally discovered from ubiquitous immune systems in prokaryotes, which endow bacteria with a unique memory against past bacteriophage infections that they utilise to mount an acquired immune response[8-9]. As a consequence of this selective pressure, an enormous diversity of different CRISPR-Cas systems exists[10]. Harnessing different interference proteins from this plethora of systems has expanded editing capabilities by providing a set of complementary proteins with different editing efficiencies across diverse organisms as well as different intrinsic specificities and requirements for targeting[11].
Employing big-data style computational pipelines to mine terabyte-scale genome sequencing data for new CRISPR-Cas systems has procured many new such systems including effector proteins used to facilitate interference. These effectors may possess novel or improvement-of-function RNA-guided catalytic activities, and form the basis of improved or alternative gene editing strategies[12-15]. This may overcome inherent issues with current gene editing tools, such as the editing efficiency at different target sites across the genome, or the introduction of large insertions and deletions as a by-product of host-cell DNA repair[8-9,16]. This review will focus on CRISPRCas system diversity to create improved or novel gene editing approaches and extend the gene editing toolbox.
The basis of CRISPR-Cas interference and cellular repair
CRISPR-Cas systems are defined as a set of genes,known as Cas associated sequences (Cas), which cooccur with a set of tandem repeat sequences, known as CRISPR[8-9]. During phage invasion in bacteria, a small fragment of DNA (protospacer), produced from phage replication is integrated into CRISPR arrays(Fig. 1)[9,17]. These arrays are structured as alternating segments of fixed-length DNA fragments, mostly acquired from previous infections (spacers) from the phage, and constant pseudo-palindromic repeat sequences (direct repeats)[9,17]. Subsequent transcription of the CRISPR array produces a series of concatenated crRNAs (pre-crRNA), which are cleaved into individual CRISPR RNA (crRNA) fragments[18].For interference, each crRNA consists of a pseudopalindromic sequence, which folds into a hairpin structure, used for recognition by the effector protein,and a guide RNA (gRNA) sequence complementary to the target nucleic acid sequence of interest (Fig. 1)[18].If a phage containing sequences present in a CRISPR array, reinvades the host bacterium, CRISPR-CascrRNA complexes use the gRNA segment of the crRNA, in conjunction with a 2 to 5 bp sequence of DNA called a protospacer adjacent motif (PAM)encoded directly adjacent to the target site, to sequence-specifically bind and cleave phage DNA or RNA (Fig. 1)[9,18-20]. In some instances, the gRNA and hairpin domains are split into two separate RNAs. The gRNA contains a crRNA and a hairpin domain with a trans-activating CRISPR RNA (tracrRNA). These two molecules form a duplex at one end of both molecules and function as a single crRNA as observed in other systems. Complementary binding then activates the cleavage activities of the effector, degrading the phage DNA and conferring immunity. This mechanism has been successfully harnessed to use the CRISPR system as a programmable nuclease to edit eukaryotic cells. CRISPR-based editing strategies rely on coercing the host-cell DNA repair pathways to integrate or substitute new genetic material[21]. Unlike other forms of DNA damage, DSBs are unique in that most organisms across the three kingdoms of life possess a homology-directed repair (HDR) pathway,which utilises a segment of DNA homologous to the severed region of the damaged DNA molecule as a template for repair[22-24]. The pathway consists of four basic steps: resection, strand invasion, re-synthesis and Holliday junction resolution[25-26](Fig. 2).
Most modern gene editing platforms leverage this pathway to modify DNA by transfecting the cells of interest with exogenous DNA homologous to the base sequence of the target site. This DNA also encodes the desired mutation to incorporate[21,27-28]. Competing with this pathway, however, is usually a second pathway known as non-homologous end joining(NHEJ), which involves the direct re-joining of the severed DNA ends without the use of a template[29-30].The accuracy of NHEJ strongly depends on the integrity of the severed DNA ends[31]. If the ends are intact, then repair is usually accurate, however if one of the ends is damaged, or the ends are incompatible,then repair usually results in the incorporation of small insertions or deletions (INDELs) at the position where the strands were severed[31]. This pathway is often used to produce loss-of-function mutations for target genes. Additionally, it is possible for a mutagenic secondary NHEJ pathway, known as microhomologymediated end joining (MMEJ) to be activated during the initial resection step of HDR[32]. This pathway attempts to re-anneal and re-join large resected regions of single stranded DNA, which always results in DNA deletions as an artefact of the repair process[32]. Which repair pathway facilitates DSB repair for a given host cell thus determines not only the type of modification induced at the target site, also the incidence of artefacts produced from the editing process that can be mutagenic to the host cell (see Burgioet al[33]for more details). In concert with technical issues related to the RNA-guided problems themselves, such as the induction of off-target breaks in other regions of the host genome, as well as limits on the efficacy and target site programmability of various effectors, this has driven a search for editing systems with novel activities that could potentially bypass the dependency of the editing platforms on DSB induction. One of the most likely sources of the building-blocks for such platforms lies in a deeper exploration of CRISPR-Cas system diversity, and unravelling the functions of as yet undiscovered or known or not yet experimentally characterised systems.
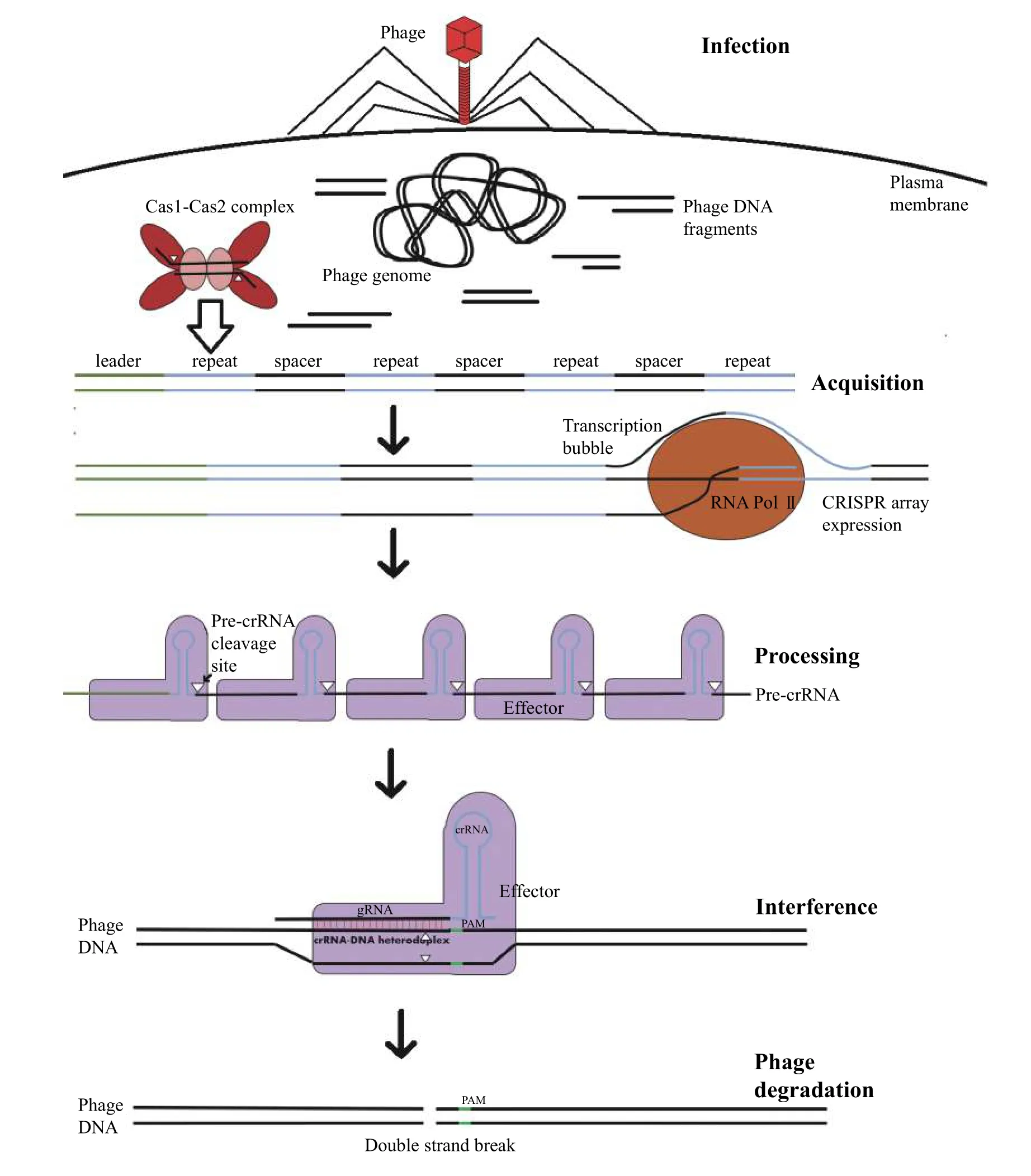
Fig. 1 Steps in CRISPR-Cas mediated acquired immunity. Although there exist countless variations in the exact proteins involved between different prokaryotic organisms, this process involves 3 distinct phases. In the adaptation phase, phage DNA fragments(protospacers) are uptaken and integrated by Cas1-Cas2 complexes into CRISPR loci. Each CRISPR is an alternating series of repeats and fragments of DNA derived from past phage infections (spacers). In response to phage reinfection, these CRISPR-arrays are then expressed and the corresponding long RNA transcripts (pre-crRNA) cleaved into individual CRISPR RNAs (crRNAs). These then complex with interference complexes, which degrade the phage genome in a sequence specific manner dictated by the spacer-derived guide RNA (gRNA)segment of the crRNA.
Diversity and evolution of CRISPR-Cas systems
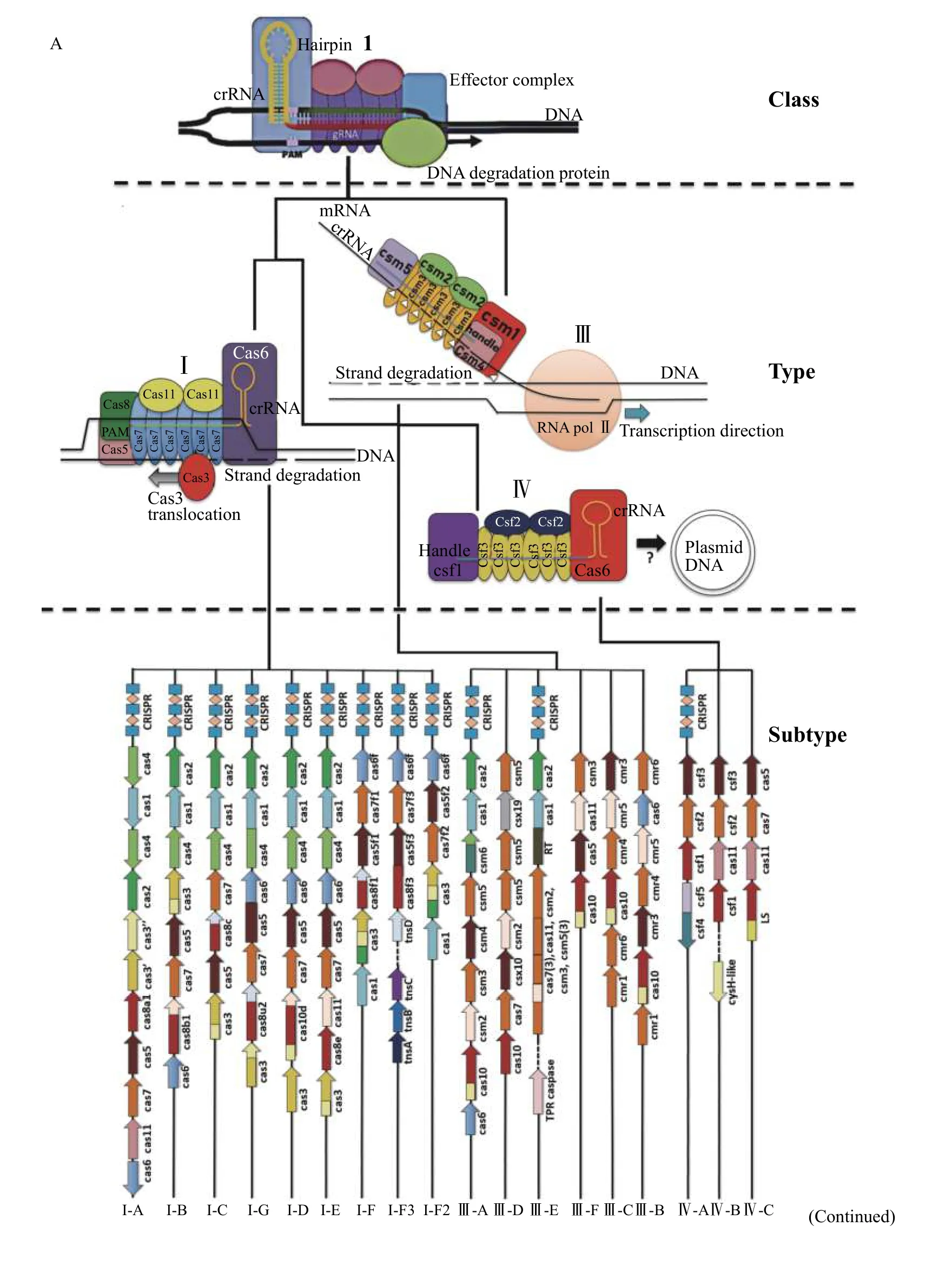
All CRISPR-Cas systems are differentiated based on the structure and function of the main effector proteins or complexes, and the presence or absence of specific CRISPR-associated proteins. Class 1 CRISPRCas systems (Fig. 3A) utilise large multi-subunit surveillance complexes, for RNA-guided DNA binding. These systems then recruit additional proteins to mediate site-specific DNA degradation[10]. In contrast, class 2 CRISPR-Cas systems all employ a single RNA-guided monomeric effector protein,which effects site-specific strand cleavage of the target nucleic acid strand (Fig. 3B)[10].
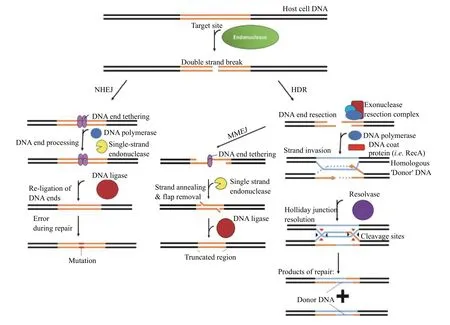
Fig. 2 Overview of the main double-stranded break repair mechanisms. There are 3 main conserved pathways available in most organisms. The NHEJ repair pathway directly tethers and re-joins the severed ends. HDR, in contrast, relies on a piece of homologous DNA(blue), which is utilised as a template for pair, and results in the incorporation of the donor DNA sequence at the break site. MMEJ occurs after the resection step of HDR, and relies on the annealing of large overhangs to re-tether the break site strands. Excision of the overhanging single-stranded DNA reseals the break but results in the deletion of DNA around the break site. NHEJ: non-homologous end joining; HDR:homology-directed repair; MMEJ: microhomology-mediated end joining.
Each of the respective CRISPR-Cas system classes are further classified based on the evolutionary lineage of the effector protein and sub-classified based on the presence or absence of genetically linked accessory proteins (Fig. 3)[10,12]. All CRISPR-Cas systems of the same type possess monomeric effector proteins, or effector protein complexes which descend from the same common ancestral variant[12]. For instance, all Cas9 proteins, which are the defining effector in typeⅡ CRISPR-Cas systems, are believed to descend from a single ancestral variant. There are three subtypes associated with the type Ⅱ systems all distinct to each other by either the presence of accessory proteins such as Cas4 or Csn2 important for spacer acquisition or interference. Type Ⅱ-A systems include the most commonly used protein for CRISPR gene editing,
Streptococcus PyogenesCas9[12].
Different types are also assigned a numeric designation based on common catalytic activities, and a potentially related (although not divergent)evolutionary origin followed by a letter designating a unique evolutionary lineage (Fig. 3)[12]. In the case of Class 1 systems with multiple subunits, a new effector type is defined if one of the subunits is novel or unrelated to the others (Fig. 3A).
Type Ⅰ-E systems, such as that found in the K12 strain ofEscherichia Coli, contain a surveillance complex called Cascade, which is comprised of five different proteins: Cas8e, Cas11, Cas7, Cas5 and Cas6[12]. By contrast, type Ⅰ-A systems comprise a surveillance complex only composed of four separate subunits, omitting Cas6 from the complex, but including an additional Cas3 protein ortholog used to mediate interference[12].
The diversity of class 2 effectors has been unravelled via big-data mining of assembled bacterial and archaeal genomes and metagenomes
With the exception of Cas12a, which was discovered inFrancisellanoviciaviadirect examination of proteins encoded proximal to the CRISPRarrays, all other Class 2 CRISPR-Cas effector proteins constituting novel systems have been discoveredviathe utilisation of computational pipelines which employ big-data filtration techniques to predict and isolate effector proteins based on their co-occurrence with conserved motifs in CRISPR-Cas systems, most notably CRISPR arrays (Fig. 4)[10,14,34-38]. The precursor step toward performing this process entails compiling a multi-terabyte sized block of assembled prokaryotic sequencing data from metagenomic and reference sequence databases. The genome data block is then scanned to identify a "seed" or "bait" motif of interest[15]. This can be the tandem repeat pattern which is conserved in CRISPR arrays, or conserved CRISPRassociated proteins such as Cas1/Cas2[14,35-36,34,39]. To avoid missing the detection of systems which employ a minimal structure, such as a single CRISPR-array in the absence of any co-occurring proteins, the union of multiple seeds can be taken, to link potential CRISPRassociated proteins to potentially any other CRISPRassociated proteins[14-15,40].
After seed identification, a 5-20 kb frame of DNA sequence upstream and downstream must be extracted and subject to open reading frame prediction to identify possible candidate CRISPR-associated proteins[15]. These proteins then need to be subject to filtering and clustering to produce putative families of CRISPR-associated proteins[15]. Finally, a "CRISPRicity" score is then calculated by finding the number of occurrences of each putative CRISPR-associated protein proximal to CRISPR-arrays and dividing this by the total number of occurrences throughout the entire block of genome sequencing data[15]. This proves that the putative CRISPR-associated proteins are linked to their proximally encoded CRISPR arrays.Subsequent analysis of the domains, family sequence diversity and properties of the individual co-occurring CRISPR-associated protein sequences has enabled the deduction of many of these CRISPR-associated proteins to be undiscovered effector proteins,which were then verified experimentally usingin vitrocleavage and PAM determination assays[15].
Limitations of CRISPR-Cas system discovery via computational pipelines
There are two main types of limitations, intrinsic and extrinsic with the computational pipelines that have been used to predict new CRISPR-Cas systems to date. The intrinsic limitations of computational pipelines are defined in this instance as the inherent flaws with the pipeline methodology itself, while the extrinsic limitations are those defined as outside the scope of the goals of previously constructed pipelines used for CRISPR-Cas system discovery.
Foremost among the intrinsic limitations is a tradeoff between the sensitivity, speed and computational resources required to compute a CRISPR-"icity"score, or equivalent, to validate that putative CRISPRassociated proteins are encoded next to CRISPRarrays[15].
Using CRISPRs or conserved CRISPR-Cas proteins as seeds comes with the logical corollary that proteins which play important roles in CRISPR-immunity, but are not genetically co-encoded with CRISPR-arrays or conserved proteins, are not detectable[41-43]. Within the members of each cluster of predicted CRISPRassociated proteins, it is assumed that similar homology between related family members implies a conserved function between all members[14,35-38,39,44].Fewer efforts to date have been made to discriminate the functionality of CRISPR-associated proteins encoded within the same families as opposed to novel families of CRISPR-Cas proteins. Prior studies of the CRISPR-Cas9 effector proteins have already unveiled immense functional diversity from within a single lineage of effector proteins, which implies that similar diversity within other CRISPR-Cas systems exists,and is not characterised by the pipelines, which identify these proteins from within genome sequencing data[45-49].
The pipeline itself also possesses several intrinsic trade-offs between the allowed sensitivity for clustering and co-occurrence score calculation, the false positive rate at which novel putative CRISPR-associated proteins are detected, and the computational requirements to perform the clustering and cooccurrence calculation steps. Because most CRISPRassociated proteins are larger than the maximum size of the short tagged reads which serve as the raw output from metagenome sequencing, only assembled metagenome data can usually be used to detect CRISPR-associated proteins using a bait-based approach[14,35-36,38,44]. Within the pipeline itself, a tradeoff exists between the false-discovery rate for CRISPR-associated proteins and the window size[15].A trade-off also exists between clustering or sequence search algorithm sensitivity and the amount of computational resources required to perform these calculations[50]. For both classes of algorithms, the underlying reason is that a more sensitive search almost always requires performing a larger number of comparisons between the decomposed query sequence(words) and the sequence database[50-52]. As a result,most pipelines have employed algorithms, such as mmseqs2, which optimise this speed/sensitivity trade-off[15,50,53].
Utilisation of the type Ⅰ systems for genome engineering
There have been several attempts to perform both gene knockout and HDR based gene editing using type Ⅰ CRISPR-Cas effectors[56]. This is possible because the single-stranded DNA degradation activity of Cas3, when applied to host-cell genomes, results in a single-stranded resected product anyway from 1 to> 50 kb in size, but most commonly in the range of around 5 to 10 kb[56], which has the potential to trigger the HDR repair pathways to incorporate foreign DNA into the host cell genome within the resected region(Fig. 5A). The key dilemma is that the single stranded progressive degradation catalytic activity of Cas3 when utilised for gene knockout results in lower efficacy in the range of 5% to 60% compared with Cas9 (10%-80%) when the DNA target site and mutation sites overlap[56-57]. Notably, however, Cas3 showed slightly improved HDR knock-in efficacy(0.8%) compared with Cas9 (0.45%) for one target site when the mutation site was significantly downstream of the target site[56].
An alternate, novel gene editing approach may come from the discovery of type Ⅰ-F systems, which naturally facilitate the programmable integration of Tn7 transposon DNA into a specific locus within a host cell (Fig. 5B). It has recently been discovered that a novel subclass of type Ⅰ and Ⅴ effectors are capable of site-specifically directing the integration of Tn7 transposons[58]with an efficiency in the range of 15% to 60% in bacteria depending on the exact target site specified[59-60]. Unfortunately, there have been no attempts to date to determine whether the use of such system is feasible in mammalian cells. Nevertheless,this represents an important advance a potentially viable alternative approach for integrating DNA into the chromosomes of bacteria.
Type Ⅲ and Ⅳ CRISPR-Cas effectors:Underutilised types with novel editing properties?
Both Type Ⅲ and Ⅳ systems employ a surveillance complex with significant structural and mechanistic similarities to type Ⅰ. In type Ⅲ systems, rather than binding directly to DNA, the complexes natively bind single stranded RNA in a sequence specific manner(Fig. 3A). Csm3 subunits are recruited to the complex upon the formation of a dsRNA duplex and introduce multiple breaks in a target RNA strand[61]. In some systems, the Cas10 subunit of the surveillance complex is able to concurrently degrade the complementary DNA strand. There have been several investigations where Type Ⅲ systems naturally present in bacteria were co-opted for gene editingviathe transformation of the target organisms with plasmid encoded CRISPR-arrays containing spacers complementary to a read-out gene of interest.However, to date, no Type Ⅲ has been expressed from vectors in the same manner as type Ⅰ systems for editing purposes, which makes it difficult to assess the potential of these systems as transferable gene editing platforms in other organisms[62-63].
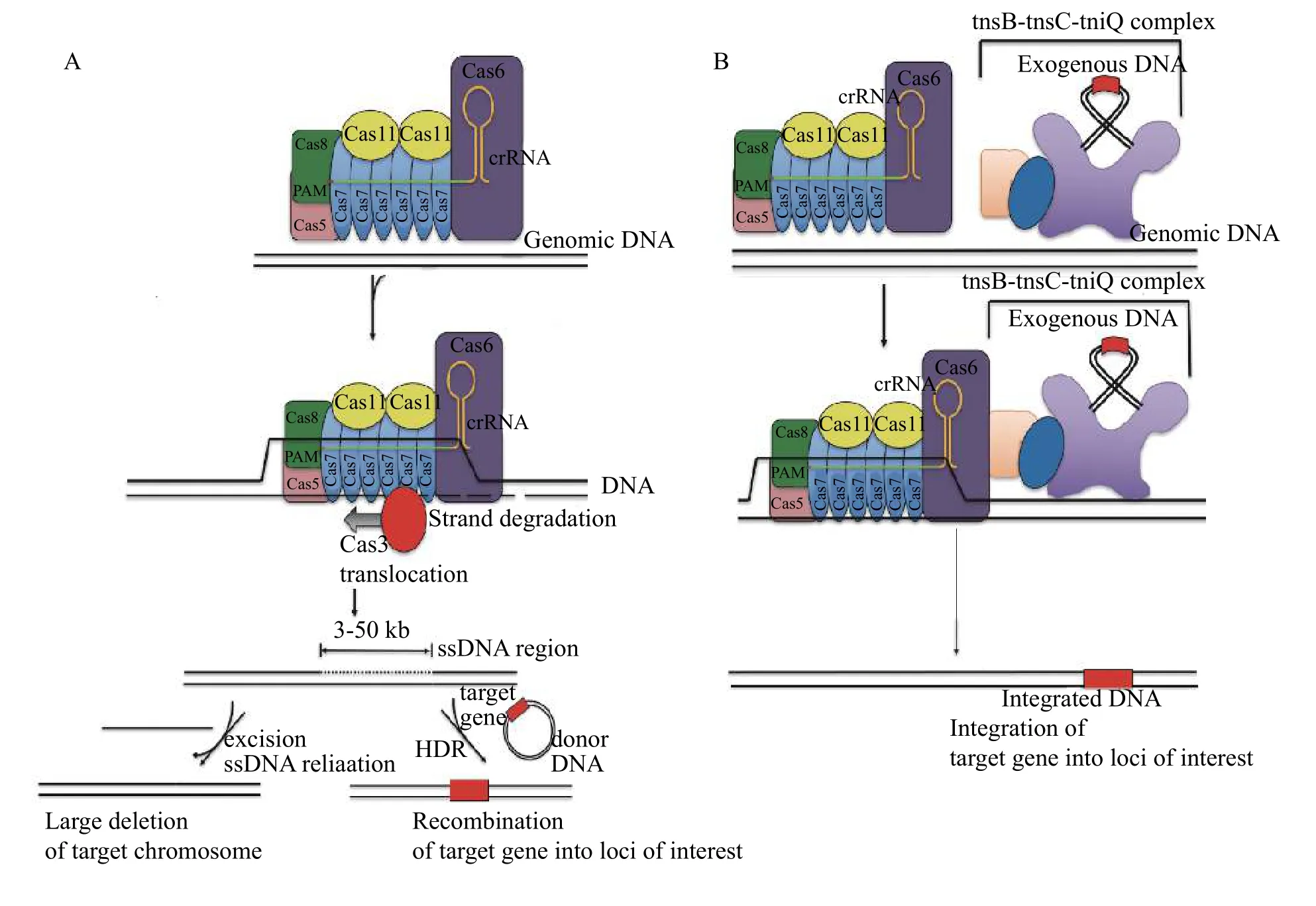
Fig. 5 Gene editing strategies developed using standalone type I systems. A: Use of the Cascade Cas3 complex to resect a large region of DNA downstream of the target enables the introduction of large deletions or gene knock-in, via either end-joining or homology-directed repair (HDR) pathways. B: A Tn7 - transposon forms a complex with a Type I-F Cascade complex to integrate DNA containing a gene of interest into the target loci.
Another potential difficulty associated with the utilisation of type Ⅲ systems is that compared with the CRISPR-Cas systems of other types, the activity of the surveillance complex appears to be more integrated with the host cell defence response[64-65].Upon target recognition, the surveillance complex synthesises cyclic oligoadenylate molecules whilst simultaneously facilitating processive single stranded DNA degradation of the transcribed gene[64,66-72].These molecules then act as signalling substrates to stimulate the binding of interference proteins to facilitate RNA strand degradation[64,70]. However, they also activate non-specific nucleases such as NucC,which is found to be co-encoded with certain type Ⅲsystems[65,73]. Upon being activated, this protein nonspecifically degrades the host genome[65]. This is a form of abortive infection to prevent the phage infection spreading to other cells in the same colony[65]. This may be an issue when utilising these proteins for gene editing, if the transformation of typeⅢ CRISPR-Cas systems into a bacterium of interest induces cell death as a side effect. This means that,although potential editing applications exist for typeⅢ systems, a significant amount of further research would be required to employ these type Ⅲ effectors in bacterial and eukaryotic cells as general purpose editing tools.
Like type Ⅰ systems, Type Ⅳ systems consist of an RNA-guided multi-subunit surveillance complex which sequence specifically binds DNA[74]. However,unlike type Ⅰ and Ⅲ, type Ⅳ systems lack Cas1-Cas2 acquisition proteins and are mainly encoded on plasmids[75]. The spacers from their corresponding CRISPR arrays also appear to map to other plasmids,rather than host cell, or phage genomes wherein they have been observed to mediate interference[74-75].Intriguingly, this appears to beviaa different mechanism to single-stranded DNA cleavage, instead requiring DinG, a protein with distant homology to helicase proteins used in recombinational repair[74].The key potential advantage of utilising type Ⅳsystems for editing, is that the interference modules consist of fewer subunits than Types Ⅰ/Ⅲ which makes them easier to compact into delivery constructs[10,74-75]. However, given that the exact mechanism interference in these has not yet been characterised[74-75], it remains unclear what, if any applications for gene editing these systems possess.
Class 2 effectors: monomeric endonucleases,which form the cornerstone of all modern gene editing toolboxes
It is widely accepted that the lion's share of gene editing advances enabled by CRISPR based gene editing have come from the discovery and utilisation of class 2 CRISPR-Cas effectors. Diverse lineages of class 2 effector proteins possess several generalisable properties, which grant them a comparative advantage in gene editing and knockdown applications compared with class 1 systems. Class 2 effector proteins are monomeric and sequence specifically degrade DNA at a single loci within the target genome or transcriptome upon binding, usually introducing either a single or double strand break in the target nucleic acid strand[10].This avoids the possible side effects arising from degrading a large DNA region as is the case in type ⅠCRISPR-Cas effector mediated gene editing[56-57]. As a consequence of these properties, the design and operation of computational pipelines, to extract and characterise the full extent of class 2 effectors diversity has been a major priority in the last 5 years.This has unveiled 3 basic types of class 2 effectors(Fig. 3B), subclassified into a plenitude of subtypes with the potential to complement or surpass the traditional first generation SpCas9-CRISPR mediated gene editing platform.
Cas9, the heart of CRISPR-Cas gene editing revolution
It is indisputable that the breakthrough generated from the utilisation ofStreptococcus pyogenesCas9(SpCas9) to induce programmable RNA-guided Double Stranded cleavage has since become the epicentre of the CRISPR-Cas gene-editing world.Cas9 distinguishes itself from other effector types by its high abundance (present in approximately 10% of bacteria[12]), distinct mechanism of double stranded cleavage (Fig. 6A), high efficacy in diverse organisms and relatively low restrictions on programmability due to small PAM requirements[76-77]. While other type Ⅴeffectors utilise a single RuvC domain to cleave both DNA strands, Cas9 uses its RuvC and HNH domains to cleave the complementary and non target strands almost simultaneously[78]. This cleavage is specific to the target site, with no indiscriminate single stranded DNA (ssDNA) cleavage occurring as a side reaction,which is often observed with the effectors of Type Ⅴsystems such as Cas12a[79].
Due to being the first ortholog discovered, there has been a much greater exploration of Cas9 ortholog diversity and re-engineering of successful orthologs into higher activity variants than for effectors from other types[80](Table 1)[27,46-47,81,101-103]. The driving motivation for using different Cas9 orthologs for editing lies in their different PAM requirements,protein size to fit into a delivery vector and editing efficacy at different target sites in different organisms.BothStaphylococcus aureusCas9 (SaCas9, 1053 residues) andCampylobacter jejuni Cas9(CjeCas9,984 residues) are smaller than SpCas9, the standard effector used for most editing applications[46,81]. This results in a smaller construct size when genes encoding either of these effector proteins are cloned onto an insertion vector for gene delivery Certain orthologs may also provide an efficacy and specificity improvement when used at certain target sites, due fewer possible off-target sites due to the effector's PAM being more specific to the target site of interest[82-83]. Although overall, there has been relatively little success in finding a naturally occurring Cas9 ortholog, which surpasses SpCas9 in terms of functionality for general purpose use, when used as a collective toolbox for a specific target site, the utilisation of these alternative Cas9 orthologs can significantly increase the specificity and efficacy of the editing reaction, as well as the number of possible sites to induce cleavage in a gene of interest[84].
Cas12 effectors facilitate staggered double strand cleavage in a manner distinct from Cas9
Although all Cas12 effectors possess and utilise a single RuvC domain for nucleic acid strand cleavage,the substrate requirements and mechanism of cleavage differ substantially between different effector types(Fig. 6B-F). To date there are 11 different known Cas12 effector types, alphabetised A to K (Fig. 3B).The mechanism underlying this cleavage has predominantly been studied in Cas12a orthologs, but provides some transferable insight into the process for other Cas12 effectors as well[85-88]. For Cas12a, RNADNA heteroduplex formation induces conformational change in the NUC lobe, making the RuvC catalytic residues accessible[88]. Cleavage of the non-target strand must precede cleavage of the target strand[89].This results in a staggered cleavage pattern with approximately 5 nt 5′ overhangs (Fig. 6B)[89]. This releases the PAM-distal target DNA fragment.However, the ribonucleoprotein complex is still catalytically competent while bound to the PAMproximal DNA[89-90]. This often results in the activation of a secondary activity wherein indiscriminate cleavage, or 'trans' cleavage of ssDNA (and in some orthologs single stranded RNA (ssRNA), and nicking of dsDNA) by the effector protein occurs[79,89].
Miniature Cas12 effectors are capable for facilitating DNA strand cleavage
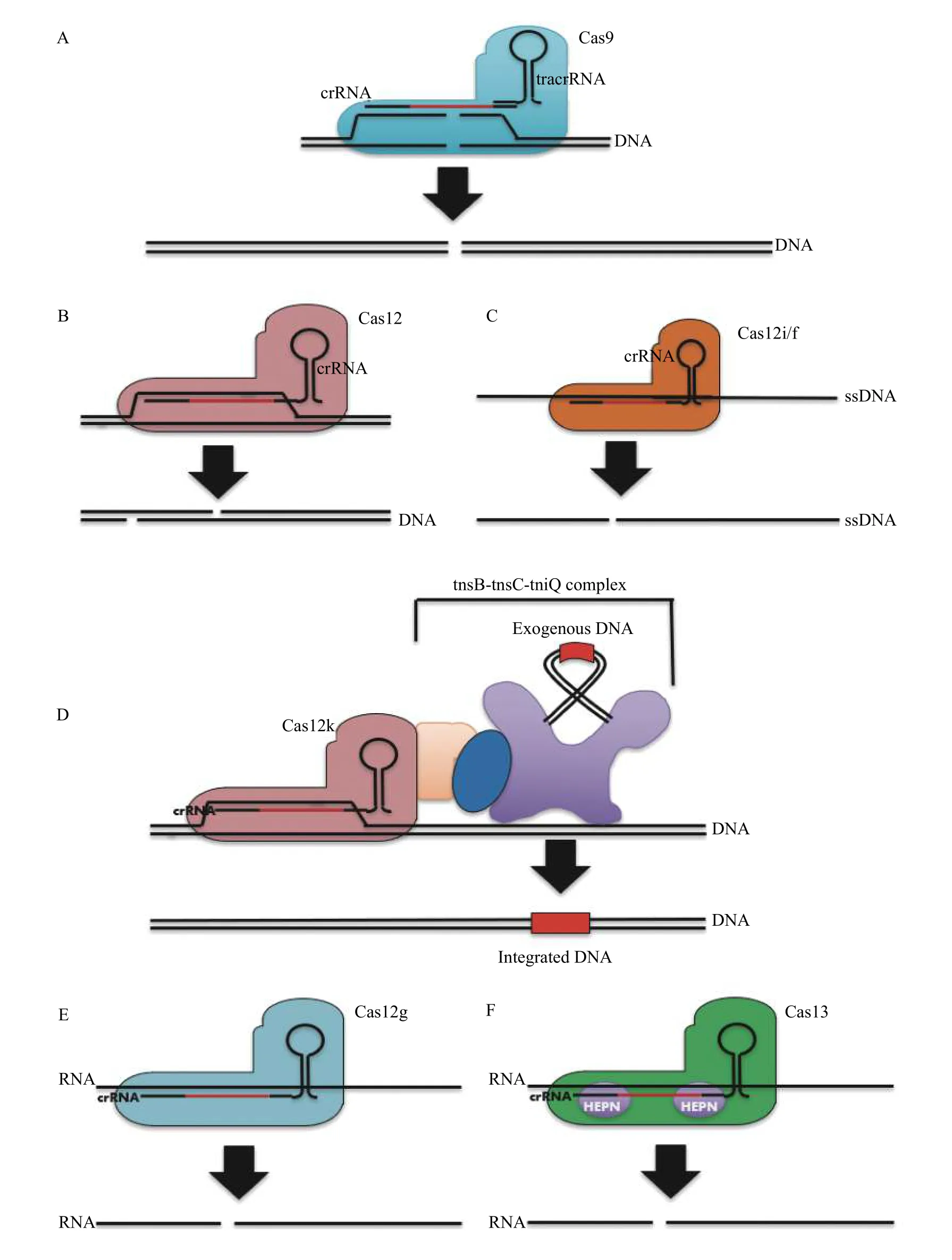
Fig. 6 Activities of Class 2 CRISPR-Cas effectors utilized in gene editing systems. A: The mostly commonly used CRISPR-Cas effector: Cas9, elicits blunt double strand cleavage via two independently functioning RuvC and HNH domains. B: This deviates from most Cas12 effector variants which induce a single staggered DSB at the target site. C: A minority of Cas12 effector types, such as Cas12i and Cas12f, most efficiently induce single stranded DNA breaks, although some representative Cas12f effectors may induce double strand breaks with limited efficacy at the target site as well. D: A special class of Cas12 effectors, designated Cas12k, are recruited by Tn7 transposons to direct the site of transposon integration in a programmable RNA-guided manner. E and F: Both Ca12g (E) and all known Cas13 effectors (F)elicit single strand cleavage of RNA, using an RuvC and HEPN domain respectively.
One recent advance has been the discovery and characterisation of Cas12j (phi) effectors. These proteins, encoded exclusively on the genomes of large phages and more compact (700 to 800 aa in size) than other Cas12 effectors[92]. While these effectors possess dsDNA cleavage activity, there was a significant difference in efficacy between Cas12j mediated cleavage of the target strand, and Cas12a mediated cleavage of the target strand[38,39,91-92]. This shortcoming means that while the characterisation of Cas12j represents an important step towards more compact,high efficacy editing proteins, it is however unlikely to supersede existing editors such as Cas12a or Cas9.
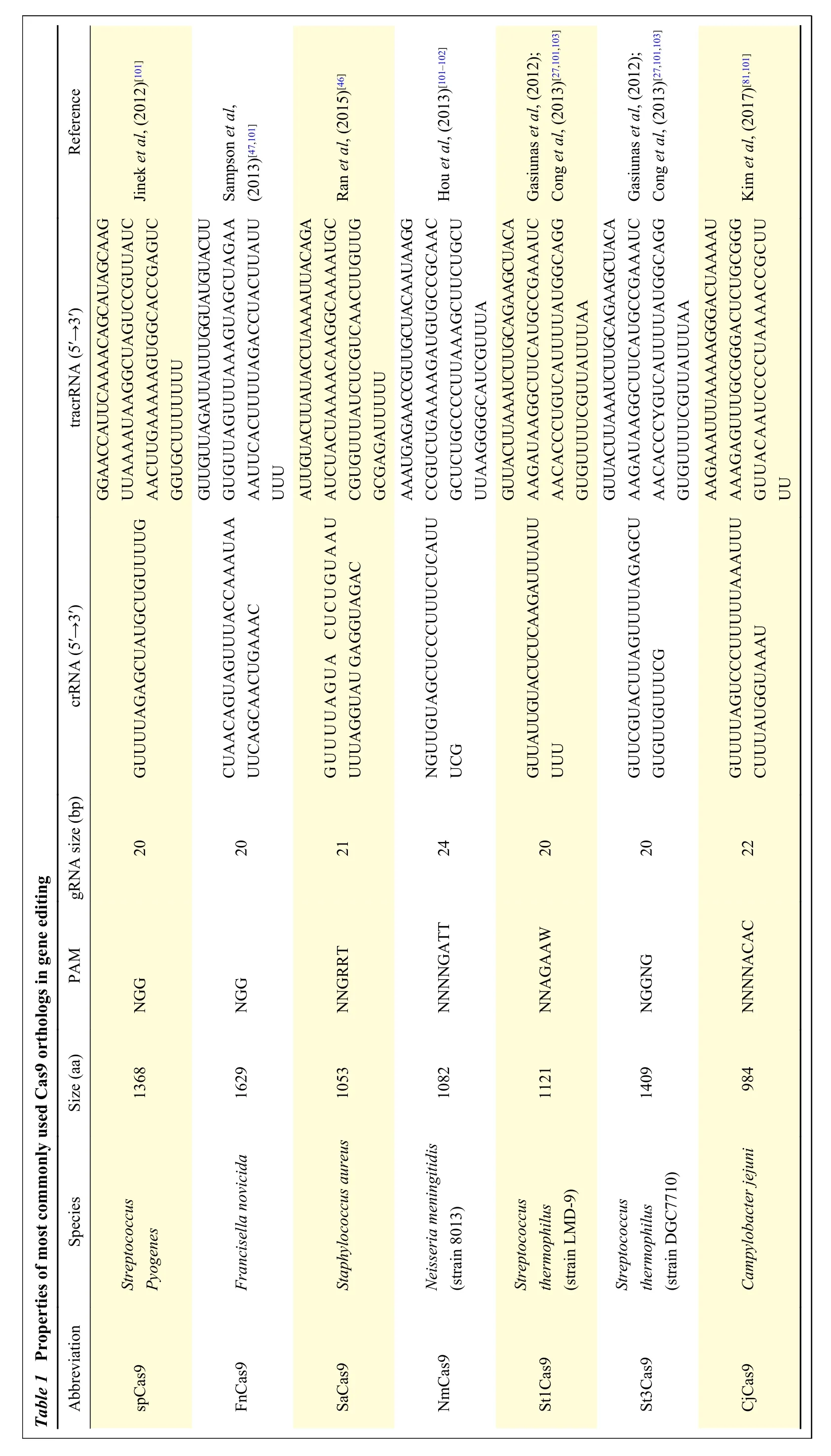
Reference Jinek et al, (2012)[101]Sampson et al,(2013)[47,101]Ran et al, (2015)[46]Hou et al, (2013)[101-102]Gasiunas et al, (2012);Cong et al, (2013)[27,101,103]Gasiunas et al, (2012);Cong et al, (2013)[27,101,103]Kim et al, (2017)[81,101]tracrRNA (5′→3′)GGAACCAUUCAAAACAGCAUAGCAAG UUAAAAUAAGGCUAGUCCGUUAUC AACUUGAAAAAGUGGCACCGAGUC GGUGCUUUUUUU GUUGUUAGAUUAUUUGGUAUGUACUU GUGUUAGUUUAAAGUAGCUAGAA AAUUCACUUUUAGACCUACUUAUU UUU AUUGUACUUAUACCUAAAAUUACAGA AUCUACUAAAACAAGGCAAAAUGC CGUGUUUAUCUCGUCAACUUGUUG GCGAGAUUUUU AAAUGAGAACCGUUGCUACAAUAAGG CCGUCUGAAAAGAUGUGCCGCAAC GCUCUGCCCCUUAAAGCUUCUGCU UUAAGGGGCAUCGUUUA GUUACUUAAAUCUUGCAGAAGCUACA AAGAUAAGGCUUCAUGCCGAAAUC AACACCCUGUCAUUUUAUGGCAGG GUGUUUUCGUUAUUUAA GUUACUUAAAUCUUGCAGAAGCUACA AAGAUAAGGCUUCAUGCCGAAAUC AACACCCYGUCAUUUUAUGGCAGG GUGUUUUCGUUAUUUAA AAGAAAUUUAAAAAGGGACUAAAAU AAAGAGUUUGCGGGACUCUGCGGG GUUACAAUCCCCUAAAACCGCUU UU crRNA (5′→3′)GUUUUAGAGCUAUGCUGUUUUG CUAACAGUAGUUUACCAAAUAA UUCAGCAACUGAAAC GUUUUAGUA CUCUGUAAU UUUAGGUAU GAGGUAGAC NGUUGUAGCUCCCUUUCUCAUU UCG GUUAUUGUACUCUCAAGAUUUAUU UUU GUUCGUACUUAGUUUUAGAGCU GUGUUGUUUCG GUUUUAGUCCCUUUUUAAAUUU CUUUAUGGUAAAU Table 1 Properties of most commonly used Cas9 orthologs in gene editing gRNA size (bp)PAM Size (aa)Species Abbreviation 20 NGG 1368 Streptococcus Pyogenes spCas9 20 NGG 1629 Francisella novicida FnCas9 21 NNGRRT 1053 Staphylococcus aureus SaCas9 24 NNNNGATT 1082 Neisseria meningitidis(strain 8013)NmCas9 20 NNAGAAW 1121 Streptococcus thermophilus(strain LMD-9)St1Cas9 20 NGGNG 1409 Streptococcus thermophilus(strain DGC7710)St3Cas9 22 NNNNACAC 984 Campylobacter jejuni CjCas9
One of the most exciting potential advances arising from the exploration of CRISPR-Cas effector diversity has been the discovery of tiny, 400 to 700 amino acids long effector proteins. These proteins are small enough to be delivered in a recombinant adenoviral vector (rAAV)[39]. Unfortunately, all Cas14 effectors discovered to date cleave ssDNA with relatively high efficacy, but are unable to cleave dsDNA with comparable efficacy which limits their potential application without protein engineering optimization(Fig. 6E)[39,91]. Nevertheless, there is considerable optimism that this limitation can be surmounted eitherviadirect protein engineering of Cas14 to produce a gain of function variant with higher cleavage activity,orviafurther exploration and characterization of Cas14 orthologs.
A small subset of Cas12k effectors mediate sitespecific integration
A subclade of Cas12 effectors exist that lack functional RuvC catalytic residues and occur in the same operon as Tn7-like transposases. These function as RNA-guided DNA binding proteins and form a complex with Tn7 transposases to direct the site of transposon integration (Fig. 6D). Compared with typeⅠ-Tn7 transposon integration systems, Cas12k guided systems offer two important distinct advantages in the form of higher insertion efficacy, depending on the loci chosen for targeting, and simpler and smaller construct size, due to the monomeric nature of Cas12k effectors compared with type Ⅰ-F and Ⅰ-B Cascade surveillance complexes[93]. The efficacy of integration by Cas12k proteins (in the range of 15% to 65% inE. Coli[60]) is superior to the measured efficacy in yeast and eukaryotes of prime editing, an alternate means of integrating DNA in host genomes[94]. This efficacy is also competitive with the editing efficacy of Cas9 or Cas12a without the side-effects associated with the utilisation of DSB repair pathways for editing, although further research is needed to demonstrate feasibility in eukaryotic cell lines[94-96].
Utilising RNA-guided gene knockdown as an alternate to gene knockout when producing null mutant strains
A different avenue for bypassing the limitations of DSB based gene knockout protocols is to induce a gene knockdown at the target site. This involves utilizing RNA-guided site-specific riboendonucleases to target and cleave mRNA transcribed by the gene of interest[14,37,38,97-98]. This silences the expression of the target gene. Several Cas12 orthologs have been discovered which possess RNA-guided RNAse activity (Fig. 6CandE), and an entire clade of CRISPR-Cas effectors, designated Cas13, have been discovered which exclusively target and cleave single strand RNA in a manner analogous to RNA-guided DNA targeting CRISPR-Cas effectors[14,37-98-99].
Concluding remarks
Exploring the diversity of CRISPR-Cas systems has unveiled a plethora of possible candidate RNA-guided ribonucleoprotein modules. However, despite the hundreds of thousands of CRISPR-Cas systems,which have been detected in metagenome sequencing data, there has been a much smaller diversity of catalytic activities that could be utilised for alternate gene editing strategies. Some of these systems,employed as emerging technologies have clear potential to become competitive with CRISPR-Cas9 in terms of editing efficacy and target site specificity,while simultaneously lacking the in-built constraints imposed by a reliance on double strand breaks to facilitate genetic recombination. Overall, when added to the existing CRISPR-toolbox, these new RNAguided interference or integrase systems represent a significant advance when used in specialised cases where the limitations CRISPR-Cas9 are a clear technical obstacle. In the near future we will be able to customise and personalise the gene editing approach by choosing an ideal nuclease for a specific application. In conjunction with other strategies such as either rational, directed evolution or the use of base-editing or prime-editing technology, the exploitation of CRISPR-Cas system diversity remains a promising avenue for developing a general purpose gene editing platform equivalent or superior to the current CRISPR-Cas9 paradigm.
Acknowledgments
We would like to thank Anthony Newman and Jovita De Silva for their efforts in proofreading this work and suggesting improvements. We would like to thank the two anonymous reviewers for their suggestions and comments that have significantly improved the manuscript. Gaetan Burgio is supported by the National Collaborative Research Infrastructure(NCRIS)viaPhenomics Australia, the National Health and Medical Research Council of Australia(Grant No. APP1143008), the Australian Research Council (Grant No. DP180101494) and the National Natural Science Foundation of China (Grant No.81772214).
杂志排行

THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Generating a CRISPR knockout mouse through a strong premature termination codon: a cautionary tale
- A transgenic pig model expressing a CMV-ZsGreen1 reporter across an extensive array of tissues
- Progress and challenges in CRISPR-mediated therapeutic genome editing for monogenic diseases
- Genome engineering technologies in rabbits
- Therapeutic gene editing strategies using CRISPR-Cas9 for the β-hemoglobinopathies
- Base editing: a brief review and a practical example